- HTAP
-
Hypertension artérielle pulmonaire
Identifiée pour la première fois en 1891 par le Docteur Ernst Von Romberg, l'hypertension artérielle pulmonaire (HTAP) correspond à un groupe de maladies d'évolution progressive caractérisée par une élévation anormale de la pression sanguine au niveau des artères pulmonaires, dont le symptôme principal est un essoufflement d'effort.
Selon la cause, l'hypertension artérielle pulmonaire peut être une maladie sévère avec une tolérance à l'effort très nettement diminuée et une insuffisance cardiaque droite.Lorsqu'aucune cause n'est déterminée, on parle alors d'hypertension artérielle pulmonaire idiopathique (dans sa forme sporadique) ou familiale ou encore d'hypertension artérielle pulmonaire primitive. Elle est parfois associée à diverses maladies (maladie auto-immune, infections,...) ou à l'utilisation de drogues (amphétamines anorexigènes...).
Il existe également diverses maladies qui s'accompagnent d'une hypertension artérielle pulmonaire, telle l'embolie pulmonaire par exemple.L'hypertension artérielle pulmonaire était initialement classée en deux catégories : la forme primitive et la forme secondaire. Une nouvelle classification, plus exhaustive, a été proposée.
La faible spécificité des symptômes entraîne souvent un retard au diagnostic.
Le traitement est médical et d'efficacité modeste.
Sommaire
Épidémiologie
La prévalence est estimée à 15 cas pour un million d'habitants (adultes) et l'incidence de 2,4 cas par millions d'habitants (adultes) par an.
Elle touche 1,7 femmes pour 1 homme. Le pic de fréquence se situe entre 30 et 40 ans.Physiopathologie
Mécanismes
La pression de l'artère pulmonaire est une pression pulsatile. On définit ainsi, comme pour la pression artérielle systémique (celle que l'on prend classiquement au bras avec un tensiomètre), une pression systolique (la plus grande) et une pression diastolique (la plus petite). On définit également une pression moyenne dont la valeur est intermédiaire des deux autres (la courbe de pression n'étant pas régulière, la pression moyenne n'est pas égale à la moyenne des pressions systoliques et diastolique).
L'hypertension artérielle pulmonaire est définie par une élévation de la pression artérielle pulmonaire (PAP) moyenne, qui devient supérieure à 25 mm de mercure au repos ou 30 mm de mercure à l'effort, alors que chez le sujet sain, elle est comprise entre 10 et 15 mm de mercure. Il existe une augmentation de la PAP lors du vieillissement, mais cette augmentation est discrète (de l'ordre de 1 mm de mercure par tranches de 10 années).
La pression artérielle pulmonaire est dépendante de la pression capillaire pulmonaire, du débit sanguin pulmonaire et des résistances vasculaires pulmonaires selon la formule suivante :
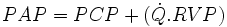
avec :
- PAP : Pression artérielle pulmonaire moyenne,
- PCP : Pression capillaire pulmonaire,
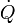 : Débit sanguin pulmonaire,
: Débit sanguin pulmonaire,- RVP : résistance vasculaire pulmonaire.
D'après cette formule on déduit le calcul:
- Résistances veineuses pulmonaires (RVP) = (PAPm (mm Hg)-PCAPm (mm Hg)x 80 )/ DC (L/mn)
[normales 90 à 120 dynes-sec-cm-5]. On parle d'hypertension artérielle pulmonaire si la résistance veineuse pulmonaire est supérieure à 300 dynes-sec-cm-5. Cette dernière peut être indexée avec le débit cardiaque et le poids et la taille du sujet (Index cardiaque) (PAPm (mm Hg) / IC (L/mn/m2) avec une valeur normale comprise entre 2 et 4 Woods). Si elle est supérieure à 10 Woods, il existe une hypertension artérielle pulmonaire.
La pression artérielle pulmonaire va donc être augmentée en cas d'augmentation :
- de la pression capillaire pulmonaire, secondaire à une augmentation de pression veineuse pulmonaire, elle-même secondaire à une insuffisance cardiaque gauche. On parle alors d'hypertension post-capillaire.
- du débit sanguin pulmonaire, dans certaines cardiopathies congénitales avec une communication entre les cavités cardiaques gauches et droites (communication inter-auriculaire ou inter-ventriculaire, persistance du canal artériel).
- des résistances vasculaires pulmonaires, principalement dans les affections pulmonaires chroniques. On parle d'hypertension pré-capillaire.
Les mécanismes physiopathologiques principaux sont une vasoconstriction, un remodelage vasculaire et des phénomènes de thrombose qui vont progressivement entraîner une augmentation des résistances vasculaires pulmonaires.
La vasoconstriction est un composant précoce de l'hypertension artérielle pulmonaire. Elle peut être l'expression d'un fonctionnement anormal des canaux potassiques ou à un dysfonctionnement de l'endothélium vasculaire (induisant la production chronique de vasodilatateurs). Le remodelage vasculaire implique toutes les couches de la paroi vasculaire et se caractérise par des modifications des tissus (visible par Histopathologie).
L'élévation de la pression artérielle a principalement un retentissement sur le ventricule droit. Lorsqu'elle est saine, cette structure a une paroi mince qui se dilate facilement. Mais l'influence de l'hypertension aboutit à une hypertrophie ventriculaire droite, puis à une insuffisance ventriculaire responsable de dyspnée d'effort (essoufflement à l'effort), d'œdèmes de membres inférieurs, d'une augmentation du volume du foie due à sa congestion (hépatomégalie). À long terme, cela peut entraîner une insuffisance cardiaque irréversible, et même la survenue d'un arrêt cardiaque brutal lors d'un effort.
Anatomo-pathologie
Des anomalies cellulaires ont été décrites dans la vascularisation pulmonaire des sujets atteints et pourraient jouer un rôle dans le développement et la progression de la maladie. Ces anomalies incluent des dysfonctionnement de l'endothélium (couche de cellules bordant l'intérieur du vaisseau sanguin) pulmonaire avec une synthèse anormale substances vasodilatatrices (monoxyde d'azote (NO), de thromboxane A2, de prostacycline et d'endothéline (Neurohormone sécrétée par l'endothélium vasculaire : endothéline-1), une altération des canaux potassiques.
De plus, des lésions se situent dans toutes les différentes structures de la paroi des vaisseaux sanguins et incluent une hypertrophie de la media (secondaire à la fois à une hypertrophie et une hyperplasie (augmentation de volume d’un tissu ou d’un organe due à une augmentation du nombre de ses cellules) des fibres musculaires lisses et une augmentation du tissu conjonctif), un épaississement de l'intima et de l'adventice (par production accrue de matrice extracellulaire) associées à des lésions plus complexes.
Causes
Les causes les plus souvent recensées d'hypertension artérielle pulmonaire incluent l'infection par le VIH (ne donnant une HTAP que dans moins d'1% des cas[1]), la sclérodermie (30% de ces derniers ont une hypertension artérielle pulmonaire[2]) et autres maladies auto-immunes, la cirrhose et l'hypertension portale, la drépanocytose, les pathologies thyroïdiennes, certaines cardiopathies congénitales, la sarcoïdose, l'histiocytose X. Elle peut également être secondaire à l'obstruction chronique des vaisseaux pulmonaires par des caillots (HTAPS post-embolique).
Certains comportements peuvent également déclencher cette variation de pression artérielle tel que la prise de certains médicaments anorexigènes, de cocaïne, de méthamphétamine, d'alcool.
Les maladies pulmonaires responsables d'un défaut d'oxygénation du sang (hypoxie) sont également des causes d'hypertension pulmonaire : la broncho-pneumopathie chronique obstructive, le syndrome de Pickwick.
Il existe des formes familiales, liées dans près des deux tiers des cas à une mutation sur le gène BMPR2 (Bone morphogenetic protein receptor type II). La pénétrance de cette mutation est à pénétrance variable : seulement 15 à 20% des sujets porteurs de la mutation génétique développeront la maladie[3]. Une autre forme familiale est dans le cadre d'une maladie de Rendu-Osler.
Lorsqu'aucune cause ne peut être identifiée, on parle d'hypertension artérielle pulmonaire primitive ou idiopathique.
Il existe une classification suivant les causes, établie en 1998 et révisée en 2004[4] où l'hypertension artérielle pulmonaire est séparée en cinq catégories, elles-mêmes divisées en sous-catégories :
- Hypertension artérielle pulmonaire :
- forme idiopathique (aucune cause retrouvée et absence d'antécédents familiaux),
- forme familiale,
- associée à une connectivite, à une cardiopathie congénitale, une hypertension portale, une infection par le VIH, à l'utilisation de drogues et toxines, autres (désordres thyroïdiens, maladie de Gaucher, hémoglobinopathies, syndromes myéloprolifératifs),
- associée à une pathologie veineuse ou capillaire (maladie veino-occlusive pulmonaire, hémangiome capillaire pulmonaire),
- hypertension pulmonaire persistante du nouveau-né,
- Hypertension veineuse pulmonaire associée à des maladies du cœur gauche :
- maladies de l'oreillette ou du ventricule gauche,
- maladies valvulaires du cœur gauche,
- Hypertension pulmonaire associée à une maladie pulmonaire et/ou une hypoxémie (broncho-pneumopathies chroniques obstructives, syndrome d'apnée du sommeil, maladies interstitielles pulmonaires, exposition chronique aux hautes altitudes),
- hypertension pulmonaire due à une maladie thrombo-embolique (par obstruction thrombo-embolique des artères pulmonaires proximales ou distales, ou par une obstruction d'origine non thrombotique (tumeur, parasite, corps étranger)),
- Divers : sarcoïdose, histiocytose X, lymphangiomatose, compression des vaisseaux pulmonaires (adénopathies, tumeur, médiastinite fibrosante).
Diagnostic
Les signes de cette pathologie n'étant pas spécifiques, il existe souvent un retard du diagnostic. Ce retard est estimé de 18 mois à 2 ans en moyenne.
Signes fonctionnels
Il n'existe pas de signes fonctionnels spécifiques.
Le sujet peut ressentir un essoufflement (dyspnée) à l'effort (qui est le signe cardinal de cette pathologie), des douleurs thoraciques (pouvant être d'allure angineuse : retro-sternales, constrictives à d'effort et/ou au repos), une toux non productive, des malaises (voire de réelles syncopes d'effort), une asthénie (grande fatigue), des palpitations, plus rarement des hémoptysies de faible abondance (présence de sang lors d'efforts de toux), une dysphonie.
Il n'y a habituellement ni orthopnée (difficulté respiratoire en position couchée), ni dyspnée paroxystique nocturne.
Signes cliniques
Il n'existe pas de signes cliniques spécifiques à cette pathologie.
L'auscultation cardiaque peut mettre en évidence une tachycardie (cœur rapide), voire une arythmie, un éclat du deuxième bruit cardiaque (B2) au niveau du foyer pulmonaire (signe quasi-constant), un souffle systolique d'insuffisance tricuspide fonctionnelle, un souffle diastolique témoignant d'une insuffisance pulmonaire.
L'auscultation pulmonaire est habituellement normale.
L'examen clinique doit rechercher des signes d'insuffisance ventriculaire droite : gros foie (Hépatomégalie), reflux hépato-jugulaire, turgescence jugulaire, œdèmes des membres inférieurs, galop droit, signe de Harzer.
Examens complémentaires
Biologie
La biologie standard est classiquement normale. Les anomalies qui peuvent cependant être retrouvées ne sont pas spécifiques : baisse du nombre de plaquettes (thrombopénie) et anémie modérées, insuffisance rénale, anomalies du bilan hépatique, taux de prothrombine spontané bas.
Une recherche d'auto-anticorps sera effectué afin de rechercher une maladie auto-immune.
Le taux d'endothéline est élevé dans le plasma et le tissu pulmonaire des patients ayant une hypertension artérielle pulmonaire. Ce taux semble proportionnel à la sévérité et au pronostic de la maladie[5].
Electrocardiogramme
L'électrocardiogramme peut montrer des signes d'hypertrophie du ventricule droit et de l'oreillette droite. Le rythme est habituellement sinusal (normal), bien que des troubles du rythme soient possibles. Un ECG normal n'élimine pas le diagnostic.
Radiographie standard
La radiologie pulmonaire est anormale dans la majorité des cas (90%). Elle va alors montrer une augmentation de la taille de la silhouette cardiaque (cardiomégalie) et une augmentation du volume du diamètre des troncs artériels pulmonaire et des artères pulmonaires proximales.
Cathétérisme cardiaque droit
L'introduction d'un long cathéter (petit tuyau souple, généralement en plastique) sous contrôle radiologique dans l'artère pulmonaire lors d'un cathétérisme cardiaque permet de mesurer les pressions et les débits.
La définition de l'hypertension artérielle pulmonaire repose sur la constatation de chiffres anormalement élevés des pressions pulmonaires. C'est une exploration invasive de l'artère pulmonaire visant à établir avec certitude l'existence d'une hypertension artérielle pulmonaire. Il est également utile au suivi évolutif de la maladie.
Le cathétérisme droit permet également d'évaluer l'évolution de la pression pulmonaire après administration de produits potentiellement vaso-dilatateurs (monoxyde d'azote, adénosine ou époprosténol permettant d'anticiper la réponse à certains médicaments.
Échographie cardiaque
L'échographie cardiaque permet de faire le diagnostic, d'évaluer la gravité et le retentissement de l'hypertension artérielle pulmonaire et de rechercher une cause. Les cavités droites (ventricule droit et oreillette droite) sont typiquement dilatées
Elle va permettre l'estimation de la pression systolique dans l'artère pulmonaire (PAPS). Cette pression artérielle systolique pulmonaire est équivalente, à peu de chose près, à la pression systolique du ventricule droit. La PAPS est estimée en mesurant la vitesse maximale V de passage du flux sanguin régurgitant au travers de la valve cardiaque tricuspide (Valve séparant le ventricule droit de l'oreillette droite) : PAPS = 4V2 + POD. La POD (pression de l'oreillette droite) est estimée en fonction de la clinique et de l'aspect échographique de la veine cave inférieure et de sa variation avec la respiration. Elle ne peut donc être calculée que s'il existe une fuite de la valve tricuspide. D'autres calculs se basent sur la courbe Doppler d'une fuite de la valve pulmonaire qui reste, cependant, inconstante.
On retient comme anormaux des chiffres de pressions systoliques de l'artère pulmonaire supérieurs à 40 mmHg.
L'échocardiographie permet de détecter une anomalie du côté du cœur gauche (défaut de contraction, problème au niveau d'une valve...) permettant de suspecter une cause cardiaque à l'hypertension artérielle pulmonaire.
Autres examens
D'autres examens pourront être réalisés :
- Un scanner thoracique avec injection d'un produit de contraste permettant de visualiser la vascularisation pulmonaire (angio-scanner), est utilisé pour rechercher une embolie pulmonaire. L'analyse du parenchyme pulmonaire est systématique.
- De même, une scintigraphie pulmonaire de perfusion (voire de ventilation/perfusion), permet de faire le diagnostic d'embolie comme cause de l'hypertension artérielle pulmonaire.
- Des explorations fonctionnelles respiratoires permettent d'évaluer la capacité respiratoire et rechercher une broncho-pneumopathie chronique obstructive, autre cause possible de la maladie.
- La réalisation d'une biopsie pulmonaire n'a pas d'intérêt établi.
Évaluation de la sévérité
La sévérité de l'hypertension artérielle est cotée selon la classification fonctionnelle OMS/NYHA (New York Heart Association) :
- Classe I : absence de limitation de l'activité physique. Les activités physiques habituelles ne provoquent pas de troubles.
- Classe II : limitation légère de l'activité physique. Asymptomatique au repos. Les activités physiques habituelles provoquent une dyspnée ou une fatigue ou des douleurs thoraciques ou des lipothymies.
- Classe III : limitation marquée de l'activité physique. Asymptomatique au repos. Un effort moins intense que l'activité de la vie quotidienne induit une dyspnée ou une fatigue.
- Classe IV : incapacité au moindre effort. Manifestations d'insuffisance cardiaque droite. La dyspnée ou la fatigue peuvent être présentes au repos. La gêne est augmentée au moindre effort.
Un moyen simple d'évaluer la gène fonctionnelle est la distance que le patient peut marcher dans un délai fixé (habituellement 6 minutes).
Prise en charge
Le traitement de la cause, lorsqu'elle est identifiée, doit être fait en première intention (traitement d'une insuffisance cardiaque, d'une embolie pulmonaire...)
Un traitement anticoagulant est recommandé dans la plupart des cas, avec une amélioration de la survie[6].
En l'absence de cause retrouvé, le traitement de base associe, selon les cas, une oxygénothérapie, des digitaliques, les diurétiques.
Les autres traitements pouvant être proposés en plus du traitement de base sont :
- l'époprosténol (prostacycline), en intra-veineux continu,
- le bosentan, inhibiteur compétitif des récepteurs de l'endothéline, traitement par voie orale,
- le sildénafil, inhibiteur de la phosphodiestérase, qui agit en augmentant le taux de NO (nitrique oxyde), puissant vaso-dilatateur,
- l'iloprost, analogue de la prostacycline, vasodilatateur etantiagrégant plaquettaire par voie inhalée.
- Les inhibiteurs calciques ne sont actif que dans moins de 10% des formes primitives[7]. La réponse à ce type de médicament peut néanmoins être testé par un test de vasodilatation au cours du cathétérisme droit.
Ces médicaments améliorent l'essoufflement et la tolérance à l'effort mais ne semblent pas toutefois être efficace en termes de survie[8].
En dernier recours, il sera éventuellement proposée une transplantation pulmonaire ou une transplantation du bloc cœur-poumons.
Évolution et complications
L'évolution naturelle de cette maladie est très variable d'un individu à l'autre. Elle conduit plus ou moins rapidement à l'insuffisance ventriculaire droite. Le décès peut survenir en quelques mois ou après plusieurs années.
Chaque personne étant différente, l'évolution dépend de la réaction au traitement : certaines personnes seront stables plusieurs années durant, tandis que d'autres nécessiteront plusieurs traitements combinés voire un traitement chirurgical si possible telles la transplantation bipulmonaire ou cardio-pulmonaire.
Une grossesse est extrêmement dangereuse durant cette maladie, avec une importante morbidité et mortalité fœto-maternelles
Notes et références
- ↑ Speich R, Jenni R, Opravil M, Pfab M, Russi EW, Primary pulmonary hypertension in HIV infection, Chest, 1991:100;1268–1271
- ↑ Chang B, Schachna L, White B, Wigley FM, Wise RA, Natural history of mild-moderate pulmonary hypertension and the risk factors for severe pulmonary hypertension in scleroderma, J Rheumatol, 2006:33;269–274
- ↑ Austin ED, Loyd JE, Genetics and mediators in pulmonary arterial hypertension, Clin Chest Med, 2007;28:43–57
- ↑ Simonneau G, Galie N, Rubin LJ et als., Clinical classification of pulmonary hypertension, J Am Coll Cardiol, 2004;43(Suppl 1):5S–12S
- ↑ Giaid A, Yanagisawa M, Langleben D et als. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension, N Engl J Med, 1993:328;1732–1739
- ↑ Johnson SR, Mehta S, Granton JT, Anticoagulation in pulmonary arterial hypertension: a qualitative systematic review, Eur Respir J, 2006;28:999–1004
- ↑ Sitbon O, Humbert M, Jais X et als. Long-term response to calcium channel blockers in idiopathic pulmonary arterial hypertension, Circulation, 2005;111:3105–3111
- ↑ Macchia A, Marchioli R, Marfisi RM, Scarano M, Levantisi G, Tavazzi L, Tognoni G, A meta-analysis of trials of pulmonary hypertension: A clinical condition looking for drugs and research methodology, Am Heart J, 2007;153:1037–1047
- (en) Cet article est partiellement issu d’une traduction de l’article en anglais : "Pulmonary hypertension".
- Rédaction Prescrire. L'hypertension artérielle pulmonaire. Grave, quand elle est symptomatique. Rev Prescrire 2004 ; 24 : 843-846.
- Société Française d'Anesthésie Réanimation. Hypertension artérielle pulmonaire. 2004 - Page visitée le 27/03/2007.
- European Society of Cardiology. Guidelines on diagnosis and treatment of pulmonary arterial hypertension. Eur Heart J 2004 ; 25 : 2243-2279.
- Chaouat A. Kraemer J.P. Canuet M. Kadaoui N. Ducolone A. Kessler R. Weitzenblum E. Hypertension pulmonaire des affections respiratoires chroniques. Press Med 2005 ; 34 : 1465-74.
- Humbert M. Sitbon O. Chaouat A. Bertocchi M. Habib G. Gressin V. Yaici A.Ayed.Y. Weitzenblum E. Cordier J.F. Chabot F. Dromer C. Pison C. Reynaud-Gaubert M. Haloun A. Laurent M. Hachulla E. Simonneau G. Pulmonary arterial hypertension in France : results from a national registry. Am J Respir Crit Care Med 2006 ; 173 : 1023-30.
Voir aussi
Articles connexes
Lien externe
 Portail de la médecine
Portail de la médecine
Catégories : Maladie de la circulation pulmonaire | Maladie cardio-vasculaire | Maladie de l'appareil respiratoire
Wikimedia Foundation. 2010.