- Drépanocytose
-
Drépanocytose Référence MIM 603903 Transmission Autosomique récessive Chromosome Chromosome 11 11p11.5 Gène β-globine Empreinte parentale Non Mutation Substitution Nombre d'allèles pathologiques S Porteur sain Atteint en forme atténuée Incidence 300 000 cas/an dans le monde. 300/an en France Nombre de cas 120 000 000 cas Diagnostic prénatal Possible Liste des maladies génétiques à gène identifié modifier 
Drépanocytose
Classification et ressources externes
Hématies normales et drépanocytaires CIM-10 D57 CIM-9 282.6 OMIM 603903 DiseasesDB 12069 MedlinePlus 000527 eMedicine med/2126 oph/490ped/2096emerg/26emerg/406 MeSH C15.378.071.141.150.150 La drépanocytose (du grec drepnos, faucille), également appelée hémoglobinose S, sicklémie, ou anémie à cellules falciformes (sickle-cell anemia en anglais), est une maladie héréditaire qui se caractérise par l'altération de l'hémoglobine, la protéine assurant le transport de l'oxygène dans le sang.
La drépanocytose n'est pas une maladie très rare. Elle est particulièrement fréquente dans les populations d'origine africaine subsaharienne, des Antilles, d'Inde, du Moyen-Orient et du bassin méditerranéen. On estime que 50 millions d'individus en sont atteints dans le monde[1]. C'est la première maladie génétique en France[2], et probablement dans le monde[3].
Sommaire
Historique
En 1904, James Herrick, médecin à Chicago, fait la première description médicale de la drépanocytose : il examine un étudiant noir âgé de 20 ans, hospitalisé pour toux et fièvre. Le sujet est faible, a des vertiges et souffre de maux de tête. Depuis un an, il ressent des palpitations et un essoufflement comme certains membres de sa famille. L’examen du sang montre que le malade est très anémique, le nombre de ses hématies n’atteignant que la moitié de la valeur normale. L’observation d’un frottis sanguin montre des hématies inhabituelles en forme de faucille ou feuille d'acanthe.
En 1949, James Neel[4] démontre que la transmission de cette maladie est mendélienne. La même année Linus Pauling montre qu'elle est due à une structure anormale de l'hémoglobine, caractérisée par une moindre solubilité. C'est ainsi la première fois qu'on découvre l'origine moléculaire d'une maladie génétique.
En 1956, le Britannique Vernon Ingram montre qu'elle est due à un remplacement d'un acide aminé dans l'hémoglobine anormale[5]. Cela a démontré pour la première fois que les gènes déterminaient la nature de chaque acide aminé dans une protéine.
En 1978, Tom Maniatis isole le gène de la bêta globine.
En 1980, Yuet Wai Kan (en) met au point un test génétique prénatal de la drépanocytose[6].
Pathogénie
Au niveau cellulaire
Les globules rouges de l'homozygote, qui ne contiennent pratiquement que de l'HbS, acquièrent ainsi la propriété de se polymériser lorsqu'ils sont désoxygénés. Ceci explique que la falciformation des hématies soit déclenchée par le manque d'oxygène dans le sang (hypoxie) :
- in vivo, dans le sang veineux et capillaire : d'où séquestration prolongée, formation de thromboses et hémolyse facile des globules rouges dans les fins capillaires où les drépanocytes ont du mal à se frayer un passage, étant donné leur forme allongée, ainsi que dans la rate où la lenteur de la circulation crée les conditions requises pour la formation des drépanocytes, qui y seront phagocytés par le SRE.
- in vitro, lors de l'examen du sang frais entre lame et lamelle, lorsqu'on aura ajouté un corps réducteur (métabisulfite).
Au niveau moléculaire
L'allèle S, est un allèle anormal du gène régissant la structure de la chaîne bêta de l'hémoglobine. Il est responsable de la synthèse de chaînes bêta, dont un acide glutamique en position 6[7] est remplacé par une valine. L'hémoglobine qui en résulte appelée HbS (hémoglobine S pour Sickle-cell disease, dénomination anglaise de la maladie) a donc la structure "alpha2ß2S". Elle se distingue de l'hémoglobine A, normale, par sa mobilité électrophorétique plus lente, mais surtout par l'insolubilité de sa forme désoxygénée, qui cristallise facilement. Cela forme des fibres longues qui vont déformer le globule rouge.
Au niveau génétique
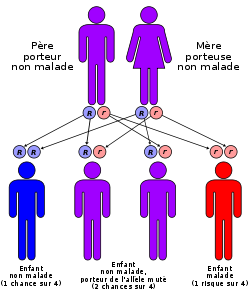 Mode de transmission
Mode de transmission
C'est le gène qui code la chaîne Béta de l'hémoglobine qui est impliqué. Ce gène est porté par le chromosome 11. La version mutée (l'allèle S) de ce gène est responsable de l'anémie falciforme. C'est un allèle co-dominant (codominance). C'est-à-dire que lorsque l'on est hétérozygote, les deux allèles s'expriment, cela pour la simple raison que chaque allèle code pour une chaîne de Beta-globine. Cependant chez cet individu, on ne constatera pas de symptômes, tout en possédant des hématies falciformes (on pourrait la considerer comme une forme atténuée).
Cette forme est appelée (S/A). La forme homozygote est la (S/S). Il s'agit de la plus douloureuse, celle dans laquelle sont décrites les crises.
Chez l'hétérozygote (S/A), les globules rouges ne contiennent pas nécessairement un mélange en proportions égales d'HbA et d'HbS.
Au niveau de l'organisme
Les globules rouges ayant perdu leur élasticité vont obstruer les capillaires provoquant une ischémie par manque d'apport d'oxygène au niveau de différents territoires. Cela explique les crises douloureuses (infarctus osseux), les infarctus cérébraux, etc. Les globules rouges pourraient également léser la paroi interne des vaisseaux (endothélium) entraînant un risque d'obstruction de ces derniers.
Ces globules rouges sont plus fragiles et vont se rompre beaucoup plus facilement, expliquant l'anémie de type hémolytique (par destruction hématies).
Épidémiologie
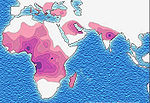 Distribution de la drépanocytose
Distribution de la drépanocytose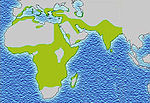 Distribution du paludisme
Distribution du paludismeSelon l'OMS, 300 000 enfants naissent avec cette maladie dans le monde chaque année[8]. En France métropolitaine, 10 000 personnes sont touchées, 2000 en Martinique et 1500 en Guadeloupe[9].
L'allèle S, responsable de l'anomalie, est surtout répandu dans le continent africain (atteignant dans certaines populations la fréquence de 30%); on le trouve également en Inde, en Arabie saoudite et dans d'autres régions du bord de la Méditerranée, en Italie (surtout en Sicile), en Grèce et en Anatolie. Les migrations ont accru la fréquence de cette maladie sur le continent américain.
Cette distribution se superpose assez bien avec celle d'une autre maladie, le paludisme ou malaria, qui, elle, a une origine infectieuse: le Plasmodium falciparum.
La présence élevée de cette maladie en Afrique semble être un cas de polymorphisme génétique équilibré entraîné par une sélection naturelle : en effet, les personnes porteuses saines hétérozygotes (A/S) ou atteintes de la drépanocytose homozygotes (S/S) sont protégées des affections neurologiques de Plasmodium, le parasite responsable du paludisme aussi appelé malaria.
Afrique
Dans certaines parties de l’Afrique subsaharienne, la drépanocytose touche jusqu’à 2 % des nouveau-nés. La fréquence du trait drépanocytaire, c'est-à-dire le pourcentage de porteurs sains qui n’ont hérité du gène mutant que d’un seul des parents, atteint 10 à 40 % en Afrique équatoriale, 1 à 2 % sur la côte de l’Afrique du Nord et moins de 1 % en Afrique du Sud. Dans les pays d’Afrique de l’Ouest (Ghana et Nigéria), la fréquence du trait drépanocytaire atteint 15 à 30 %. En Ouganda, cette fréquence atteint 45 % chez les Baambas[10].
Europe
Dans plusieurs pays ou régions d'Europe (Sud de l'Italie, Grèce, Sud du Portugal, Albanie, Sud de la Turquie), la drépanocytose est indigène avec des fréquences de porteurs du trait entre 1 et 5 % de la population. Dans d’autres pays européens (Royaume-Uni, France, Belgique, Allemagne) la maladie est apparue avec les flux migratoires en provenance d'Afrique, du Moyen Orient et d'Asie.
France
En France, les premiers cas de thalassémie majeure et de drépanocytose ont été rapportés dans les années 1940[11]. Aujourd’hui, la drépanocytose est la maladie génétique la plus fréquente en France avec plus de trois fois le nombre d’enfants atteints de mucoviscidose. On estime actuellement à 10 000 le nombre de sujets atteints de syndromes drépanocytaires majeurs (SDM), avec en France métropolitaine un nombre supérieur à celui de l’outre-mer. Chaque année, naissent en France 300 à 350 enfants atteints de SDM dont la plupart en Ile-de-France[12]. Contrairement à d'autre maladies génétiques telles la phénylcétonurie et la mucoviscidose, le dépistage de la drépanocytose en France métropolitaine n'est pas systématique mais est effectué uniquement chez les nouveau-nés dont les parents appartiennent à un groupe à risque pour cette maladie (essentiellement Afrique, Antilles et Maghreb). En 2005, sur un total de 774 355 naissances en métropole, 198 065 enfants de couples « à risque » (soit 25,6 % du nombre total de naissances en métropole) ont bénéficié du dépistage de la drépanocytose et au total, 277 syndromes drépanocytaire majeur (SDM) ont été repérés. L’incidence moyenne de la drépanocytose en métropole est de 1/715 nouveau-nés testés et de 1/2 795 sur l’ensemble des nouveau-nés[13]. En 2007, en métropole, 28,45% des nouveau-nés sont nés de couples « à risque » et ont fait l'objet de dépistages ; ce pourcentage variant selon les régions de 4,40% en Bretagne à 55,68 % en Île-de-France à raison des différences d’origine dans la population parentale. Parmi les nouveau-nés atteints de SDM recensés depuis le début du dépistage, mis en place à des dates différentes avant la généralisation à l’ensemble du territoire en 2000, la grande majorité est née en Ile-de-France, les autres enfants drépanocytaires sont nés, à proximité des grands centres urbains, principalement dans l’est et le sud du pays[14].
Région % de nouveau-nés à risque par
rapport au total des naissances (2007)Île-de-France 55,68 Provence-Alpes-Côte d'Azur 41,91 Languedoc-Roussillon 34,78 Alsace 29,29 Midi-Pyrénées 27,77 Rhône-Alpes-Pays de Savoie 27,67 Picardie 19,92 Franche-Comté 17,90 Bourgogne 17,01 Lorraine 16,14 Champagne-Ardenne 15,36 Limousin 15,16 Nord-Pas-de-Calais 14,27 Centre Val de Loire 14,03 Auvergne 12,84 Aquitaine 12,29 Normandie 11,61 Pays de la Loire/Poitou-Charentes 11,20 Bretagne 4,40 France métropolitaine 28,45 Belgique
En Belgique, l’incidence moyenne de la drépanocytose est de 1/1 710 sur l’ensemble des nouveau-nés à Bruxelles et de 1/943 à Liège.
Allemagne
Seuls 300 cas de syndromes drépanocytaires majeurs sont recensés[15].
Symptômes
L'affection se signale chez le nourrisson, mais n'est d'ordinaire pas manifeste à la naissance parce que les globules rouges du nouveau-né contiennent encore 50-90% d'hémoglobine fœtale. Les symptômes de cette maladie peuvent apparaître dès l'âge de deux à trois mois, date d'apparition de la chaîne Béta.
Les manifestations aiguës habituelles de la drépanocytose sont de trois ordres :
- Crises vaso-occlusives : des caillots bouchent une artère, entraînant des infarctus tissulaires pouvant toucher différents partie du corps (os, abdomen, rein, cerveau, rétine...). Ces crises peuvent être très douloureuses.
- Anémie hémolytique : les globules rouges des drépanocytaires sont de forme anormale, elles sont détruites au niveau de la rate.
- Infections : elles sont plus fréquentes chez les drépanocytaires, surtout à pneumocoques [16] ou méningocoque liée à la destruction de la rate par infarctus tissulaires répétés, on parle d'asplénie fonctionnelle. Elles peuvent aussi aggraver l'anémie en cas d'infection par le parvovirus B19.
Les manifestations chroniques de la drépanocytose associent un retard de taille et de poids, des déficits nutritionnels (en folates, car cette vitamine est indispensable à la création des hématies qui sont renouvelées très rapidement lors des crises d'anémie, épuisant ainsi le stock de folates), un retard pubertaire fréquent, des troubles cardio-pulmonaires (augmentation de la taille du cœur, insuffisance respiratoire), une rate augmentée de volume (mais qui avec le temps va régresser jusqu'à devenir de petite taille par atrophie), des anomalies rétiniennes (hémorragies), etc.
Hématies d'un drépanocytaireImage prise au microscope électronique du sang d'un patient hétérozygote pour la drépanocytose, sur laquelle on distingue des globules rouges sains, circulaires et concaves, des globules rouges malades possédant de l'hémoglobine mutée S, de la forme d'une faucille.
Examens et diagnostic
À part les constatations communes à toutes les anémies hémolytiques, le diagnostic repose sur la mise en évidence de l'hémoglobine S. Ceci peut se faire :
- par l'observation au microscope de sang frais conservé entre lame et lamelle (frottis sanguin), avec ou sans addition d'un réducteur (métabisulfite de potassium, acide ascorbique) : Globules rouges en faux ou feuille de houx dont la proportion dépend de la nature homozygote ou hétérozygote
- Electrophorèse de l'hémoglobine qui montrera, chez l'homozygote, une bande unique d'une hémoglobine migrant anormalement lentement, et chez l'hétérozygote la présence de deux bandes d'hémoglobine, dont la plus rapide sera l'hémoglobine A et l'autre l'hémoglobine S.
- Aujourd'hui des tests permettent de dépister les porteurs sains (personnes hétérozygotes qui possèdent l'allèle responsable mais qui ne sont pas malades) ; ils sont alors informés que l'enfant conçu par deux porteurs sains présente un risque sur quatre d'être atteint d'anémie falciforme.
Dans les pays industrialisés, le diagnostic se fait en période néo-natale si les parents sont à risque ou atteints. Dans les pays non industrialisés, le diagnostic se fait souvent à la première manifestation ou complication. Le dépistage néo natal pourrait se traduire par une amélioration du pronostic[17].
Pronostic
La maladie peut se compliquer d'infarctus cérébraux dont la prévalence atteint près de 10% des patients âgés de moins de 20 ans[18]. L'hypertension artérielle pulmonaire est une complication fréquente et grave[19]. Elle est retrouvée en échocardiographie dans près d'un tiers des cas mais cet examen s'avère peu spécifique et la prévalence réelle (confirmée par la mesure directe de la pression pulmonaire par cathétérisme cardiaque) serait bien moindre[20].
Avant l'apparition de l'hydroxyurée près de la moitié des malades décédait avant l'âge de 50 ans, soit au cours d'une crise, ou d'un accident vasculaire cérébral[21].
Traitements
Le traitement de la drépanocytose repose sur :
- le traitement des crises vaso-occlusives : antalgiques (pouvant aller jusqu'aux opiacés) et mise sous oxygène ; il est à noter que le métabolisme de la morphine est accéléré (élimination de 3 à 10 fois plus rapide que chez les sujets indemnes) chez les sujets porteurs de la maladie[22] ;
- la prévention des facteurs déclenchant les crises (froid, altitude, infections, déshydratation) ;
- la supplémentation en folates ;
- le traitement préventif des infections à pneumocoque et méningocoque ;
- la transfusion sanguine en cas d'anémie profonde ou d'infection grave.
- la transfusion-saignée permettant de réduire la proportion d'hémoglobine S
L'hydroxyurée permet de favoriser la production d'hémoglobine fœtale, formée habituellement en petite quantité et parfaitement fonctionnelle, en inhibant la production d'hématies contenant l'hémoglobine S[23]. Ce médicament semble réduire significativement le nombre de crises douloureuses et la mortalité de la maladie[24]. Il ne peut cependant pas être utilisé, de par son mécanisme d'action, chez les patients anémiques. La surveillance des paramètres sanguins doit donc être très soigneuse. L'autre obstacle à son utilisation reste son coût, qui peut toutefois générer des économies de prise en charge dans les pays économiquement développés[25].
La prévention des infections par le pneumocoque chez le jeune enfant est faite par la vaccination[26].
Des transfusions sanguines pourraient diminuer sensiblement le risque d'accidents vasculaire-cérébraux chez certains enfants particulièrement à risque (anomalie du doppler trans-crânien)[27].
Mesures simples de prévention
Pour éviter les crises il est recommandé de suivre des mesures simples suivantes:
- boire fréquemment de l'eau
- bien aérer les pièces, afin de bien s'oxygéner[réf. nécessaire]
- rester au chaud
- ne pas prendre de poids
- manger des aliments riches en fer, ou qui facilitent l'assimilation du fer (viande rouge, pâté de foie, …)
- ne pas s'enrhumer ou plus généralement éviter au maximum les infections respiratoires
- porter des vêtements qui ne coupent pas la circulation sanguine, c'est-à-dire amples
- ne pas s'essouffler
- éviter d'aller à plus de 1 500 mètres d'altitude
- ne pas s'exposer a de fortes chaleurs (la déshydratation déclenche des crises par augmentation de la viscosité sanguine[28]).
Si on se plie à ces précautions, on peut être atteint de drépanocytose très longtemps sans être affecté, même en étant sportif de haut niveau : en mai 2010, le joueur de football Lassana Diarra a dû déclarer forfait après que la maladie se soit révélée par des douleurs intestinales durant un séjour à 3 000 m d'altitude pendant un stage à Tignes, avec l'équipe de France.
Greffe de moelle osseuse
Les hématies sont produites à partir de cellules souches dans la moelle osseuse. En détruisant la moelle osseuse du malade et en la remplaçant par celle d'un donneur, il y a possibilité d'obtenir une guérison totale. Environ 200 greffes ont été réalisées dans le monde chez des drépanocytaires, permettant d'obtenir la guérison dans 85 % des cas[29]. Il faut cependant un donneur apparenté le plus possible: un frère ou une sœur. Il y a la possibilité pour les parents de recourir à une fécondation in vitro avec sélection par DPI d'embryons compatibles pour la greffe. Cette voie de traitement dite du « bébé médicament » est très encadrée par les lois de bioéthique.
Article connexe : Transplantation de moelle osseuse.Voies de recherche
Des souris drépanocytaires ont pu être guéries en introduisant chez ces animaux un gène produisant une hémoglobine "anti-drépanocytaire" en quantité élevée[30].
En 2007, le VK500 est proposé dans le traitement de la drépanocytose. Cependant, aucune étude clinique sérieuse n'a prouvé l'efficacité réelle de ce médicament[31].
Liens externes
- (fr) Association d'Anémie Falciforme du Québec
- (en) NCBI (NIH): Bibliographie sur Medline (en anglais) ;
- (fr) Orphanet: Informations sur la drépanocytose (consultations, recherches, associations, …) ;
- (fr) Rapport de l'OMS (2006).
- (fr) OILD: Organisation Internationale de Lutte contre la Drépanocytose
Références
- Arnal C. et Girot R. Drépanocytose chez l'adulte. Encycl Méd Chir (Éditions Scientifiques et Médicales Elsevier SAS, Paris), Hématologie, 13-006-D-16, 2002, 15 p.
- Conférence de Canal U
- Association drepavie
- James Neel, The inheritance of sickle cell anemia, Science vol.110, 1949, Page 543-548
- Ingram, V.M. (1956). A Specific Chemical Difference between Globins of Normal and Sickle-cell Anemia Hemoglobins. Nature 178: 792-794.
- Site de la Fondation Louis Jeantet
- Lehninger - Principles of Biochemistry
- OMS 2006
- www.drepaction.org
- Rapport de l'OMS sur la Drépanocytose, 24 avril 2006
- Lena-Russo, D ; North, M.L. etGirot, R. Épidémiologie des maladies génétiques de l’hémoglobine en France Métropolitaine. Rev Prat 1992 ; 42: 1867-72. Bardakdjian, Josiane et Wajcman, Henri Épidémiologie de la drépanocytose, La Revue du Praticien, 2004
- Montalembert M., Échanges érythrocytaires chez les patients drépanocytaires. Hématologie 2007, 13(4) : 243-9.
- Josiane Bardakdjian-Michau, Le dépistage néonatal de la drépanocytose en France, Médecine thérapeutique / Pédiatrie. Volume 11, Numéro 1, 5-8, janvier-février 2008, Dossier
- Bardakdjian-Michau, M Bahuau, D Hurtrel, et al.2008, Neonatal screening for sickle cell disease in France, J Clin Pathol 2009 62: 31-33, doi:10.1136/jcp.2008.058867
- Dickerhoff R, von Ruecker A. Manifestations of sickle cell disease in adolescents and young adults. Clinical aspects and therapy references. Klin Padiatr 1998 ; 210 : 10-6.
- de Montalembert M, Management of sickle cell disease, BMJ, 2008;337:a1397
- Vichinsky E, Hurst D, Earles A, Kleman K, Lubin B, Newborn screening for sickle cell disease: effect on mortality, Pediatrics, 1998;81:749-54
- Balkaran B, Char G, Morris JS, Thomas PW, Serjeant BE, Serjeant GR, Stroke in a cohort of patients with homozygous sickle cell disease, J Pediatr,1992;120:360
- Gladwin MT, Sachdev V, Jison ML et als. Pulmonary hypertension as a risk factor for death in patients with sickle cell disease, N Engl J Med, 2004;350:886-95
- Parent F, Bachir D, Inamo J et al. A hemodynamic study of pulmonary hypertension in sickle cell disease, N Engl J Med, 2011;365:44-53
- Platt OS, Brambilla DJ, Rosse WF, et als. Mortality in sickle cell disease: life expectancy and risk factors for early death, N Engl J Med, 1994;330:1639-1644
- (en) Recours à de haute doses de morphine chez les porteurs de la drépanocytose lire en ligne
- Platt OS, Orkin SH, Dover G, Beardsley GP, Miller B, Nathan DG, Hydroxyurea enhances fetal hemoglobin production in sickle cell anemia, J Clin Invest, 1984;74:652-656
- Steinberg MH, Barton F, Castro O, et als. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment, JAMA, 2003;289:1645-1651
- Moore RD, Charache S, Terrin ML, Barton FB, Ballas SK, Cost-effectiveness of hydroxyurea in sickle cell anemia, Am J Hematol, 2000;64:26-31
- Davies EG, Riddington C, Lottenberg R, Dower N, Pneumococcal vaccines for sickle cell disease, Cochrane Database Syst Rev, 2004;(1):CD003885.pub2
- Adams RJ, McKie VC, Hsu L, Files B et als. Prevention of a first stroke by transfusions in children with sickle cell anemia and abnormal results on transcranial Doppler ultrasonography, N Engl J Med, 1998;339:5-11
- La prévention des crises sur le site de l'Association pour l'Information et la Prévention de la Drépanocytose
- Site des hôpitaux de Lyon, consulté le 30 avril 2008]
- Pawliuk R, Westerman KA, Fabry ME et Als. [2001: Vol. 294. no. 5550, pp. 2368 - 2371 Correction of sickle cell disease in transgenic mouse models by gene therapy], Science, 2001:294;2368-2371
- Noir&Blanc Escroquerie : le VK500


Wikimedia Foundation. 2010.