- Théorie de la perturbation (chimie quantique)
-
Théorie de la perturbation (mécanique quantique)
En mécanique quantique, la théorie de la perturbation (ou théorie des perturbations) est un ensemble de schémas d'approximations liée à une perturbation mathématique utilisée pour décrire un système quantique complexe de façon simplifiée. L'idée est de partir d'un système simple et d'appliquer graduellement un hamiltonien « perturbant » qui représente un écart léger par rapport à l'équilibre du système (perturbation). Si la perturbation n'est pas trop importante, les différentes quantités physiques associées avec le système perturbé (comme ses niveaux d'énergie et états propres) seront générés de manière continue à partir de ceux du système simple. On peut donc par conséquent étudier le premier à partir des connaissances sur le dernier.
Sommaire
Application de la théorie de la perturbation
La théorie de la perturbation est un outil important pour la description des systèmes quantiques réels, car trouver des solutions exactes à l'équation de Schrödinger pour des hamiltoniens de systèmes même modérément complexes peut être très difficile. Les hamiltoniens pour lesquels on connaît des solutions exactes, comme ceux de l'atome d'hydrogène, de l'oscillateur harmonique quantique et de la particule dans une boîte, sont trop idéalisés pour décrire de manière adéquate la plupart des systèmes. Ils ne visent en particulier que des systèmes à une seule particule. En utilisant la théorie de la perturbation, on peut utiliser les solutions connues de ces hamiltoniens simples comme premières approximations de solutions pour une série de systèmes plus complexes. Ainsi, en ajoutant un potentiel électrique perturbateur au modèle quantique de l'atome d'hydrogène, on peut calculer les déplacements faibles des raies spectrales de l'hydrogène en raison de la présence d'un champ électrique (effet Stark). Ceci ne peut être qu'une approximation : la somme d'un potentiel coulombien avec un potentiel linéaire est instable bien que le temps tunnel (radioactivité) soit très important. Cela montre que même pour un ajustement des raies d'énergie spectrale, la théorie de la perturbation échoue à traduire entièrement le phénomène.
Les modifications de formules induites par l'introduction de la perturbation ne sont pas exactes, mais peuvent conduire à des résultats précis tant que le paramètre de développement reste faible. Au-delà d'un certain ordre
reste faible. Au-delà d'un certain ordre 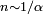 , cependant, les résultats deviennent erratiques dans la mesure où les séries générées divergent, leur développement étant seulement asymptotique. Il existe des manières de les convertir en séries convergentes, ce qui peut être considéré pour des paramètres de développement importants, en particulier par une méthode de perturbation variationnelle.
, cependant, les résultats deviennent erratiques dans la mesure où les séries générées divergent, leur développement étant seulement asymptotique. Il existe des manières de les convertir en séries convergentes, ce qui peut être considéré pour des paramètres de développement importants, en particulier par une méthode de perturbation variationnelle.
En électrodynamique quantique (QED) dans laquelle l'interaction électron-photon est traitée de manière perturbative, le calcul du moment magnétique électronique est en accord avec l'expérience jusqu'à 11 décimales, ce qui correspond d'ailleurs à la précision des mesures actuelles. Dans cette théorie, ainsi que pour d'autres théories quantiques des champs, des techniques spécifiques de calcul connues sous le nom de diagrammes de Feynman sont employées pour effectuer une somme systématique des termes de puissance donnée de la série.Dans certaines circonstances, la théorie de la perturbation est une approche invalide. Cela se produit lorsque le système à décrire ne peut être approché par une petite perturbation imposée à un système simple. En chromodynamique quantique, par exemple, l'interaction des quarks avec le champ gluonique ne peut être traité par perturbation aux faibles énergies car la constante de couplage (paramètre de développement) devient trop important. La théorie de la perturbation échoue aussi à décrire des états générés de manière diabatique à partir du « modèle libre », comme les états liants et différents phénomènes collectifs comme les solitons. On peut considérer, par exemple, un système de particules libres (non-interagissantes), dans lequel une interaction attractive est introduite. Selon la forme de cette interaction, un ensemble entièrement nouveau d'états propres correspondant à des groupes de particules liées entre elles peut être créé. Un exemple de ce phénomène peut être trouvé en supraconductivité conventionnelle, dans laquelle l'attraction portée par les phonons entre les électrons de conduction mène à la formation de paires d'électrons corrélés connues sous le nom de paires de Cooper. Lorsque l'on a affaire à de tels systèmes, on utilise habituellement d'autres schémas d'approximation, comme la méthode variationnelle ou l'approximation BKW. En effet, il n'existe pas d'analogue à une particule liante dans le modèle non perturbé et l'énergie d'un soliton varie typiquement comme l'inverse du paramètre de développement. Cependant, si l'on « intègre » sur le phénomène solitonique, les corrections non perturbatives seront dans ce cas faibles, de l'ordre de
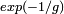 ou
ou 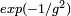 dans le paramètre de perturbation
dans le paramètre de perturbation  . La théorie de la perturbation peut seulement conduire à des solutions « proches » de la solution non-perturbée, même s'il existe d'autres solutions (qui augmentent typiquement quand le paramètre de développement approche de zéro).
. La théorie de la perturbation peut seulement conduire à des solutions « proches » de la solution non-perturbée, même s'il existe d'autres solutions (qui augmentent typiquement quand le paramètre de développement approche de zéro).
Le traitement des problèmes des systèmes non-perturbatifs a été en partie aidé par l'essor des ordinateurs modernes. Il est devenu (relativement) simple de trouver des solutions non-perturbatives pour certains problèmes, par le biais de méthodes comme la théorie de la fonctionnelle de la densité. Ces avancées ont particulièrement profité à la chimie quantique. Les ordinateurs ont également été employés pour procéder à des calculs en théorie de la perturbation atteignant de très hauts niveaux de précision, nécessaires et importants en physique des particules pour obtenir des résultats théoriques comparables à l'expérience.Théorie de la perturbation indépendante du temps
Il y a deux catégories de théorie de la perturbation : indépendante du temps et dépendante du temps. Dans cette section, on traitera de la théorie de la perturbation indépendante du temps, dans laquelle le hamiltonien de perturbation est statique. La théorie de la perturbation indépendante du temps fut présentée dans un article[1] d'Erwin Schrödinger de 1926, peu après qu'il eut énoncé ses théories en mécanique ondulatoire. Dans cet article, Erwin Schrödinger faisait référence à un travail antérieur de lord Rayleigh[2] qui étudia les vibrations harmoniques d'une corde perturbée par des petites inhomogénéités. C'est pourquoi cette théorie de la perturbation est parfois appelée théorie de la perturbation de Rayleigh-Schrödinger.
Corrections du premier ordre
On commence en utilisant un hamiltonien non perturbé
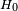 , qui est aussi considéré comme indépendant du temps. Il possède des niveaux d'énergie et états propres connus, déterminés par l'équation de Schrödinger indépendante du temps :
, qui est aussi considéré comme indépendant du temps. Il possède des niveaux d'énergie et états propres connus, déterminés par l'équation de Schrödinger indépendante du temps :Pour simplifier, on postule que les énergies sont discrètes. Les exposants
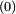 indiquent que ces quantités sont associées au système non perturbé.
indiquent que ces quantités sont associées au système non perturbé.On peut alors introduire une perturbation dans le hamiltonien. Soit
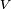 un hamiltonien représentant une petite perturbation physique, comme un potentiel énergétique produisant un champ externe (donc est formellement un opérateur hermitique). Soit
un hamiltonien représentant une petite perturbation physique, comme un potentiel énergétique produisant un champ externe (donc est formellement un opérateur hermitique). Soit  un paramètre sans dimension pouvant prendre des valeurs allant continûment de 0 (pas de perturbation) à 1 (perturbation totale). Le hamiltonien perturbé est :
un paramètre sans dimension pouvant prendre des valeurs allant continûment de 0 (pas de perturbation) à 1 (perturbation totale). Le hamiltonien perturbé est :Les niveaux d'énergie et états propres du hamiltonien perturbé sont de nouveau donnés par l'équation de Schrödinger :
Le but est alors d'exprimer
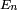 et
et 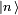 en termes de niveaux d'énergie et d'états propres de l'ancien hamiltonien. Si la perturbation est suffisamment faible, on peut les écrire en séries entières de :
en termes de niveaux d'énergie et d'états propres de l'ancien hamiltonien. Si la perturbation est suffisamment faible, on peut les écrire en séries entières de :où
et
Lorsque
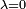 , les équations se réduisent à celles non perturbées, qui sont les premiers termes de chaque série. Lorsque la perturbation est faible, les niveaux d'énergie et les états propres ne devraient pas beaucoup différer de leurs valeurs non perturbées, et les termes de perturbation devraient rapidement devenir plus petits au fur et à mesure que l'ordre augmente.
, les équations se réduisent à celles non perturbées, qui sont les premiers termes de chaque série. Lorsque la perturbation est faible, les niveaux d'énergie et les états propres ne devraient pas beaucoup différer de leurs valeurs non perturbées, et les termes de perturbation devraient rapidement devenir plus petits au fur et à mesure que l'ordre augmente.
Si l'on introduit ces séries dans l'équation de Schrödinger, on obtient :Le développement de cette équation et la comparaison des coefficients de chaque puissance de
conduit à un système d'équations infini. L'équation d'ordre 0 est tout simplement l'équation de Schrödinger du système non perturbé. L'équation au premier ordre est :que l'on multiplie par
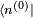 . Le premier terme de gauche s'annule avec le premier terme de droite (le hamiltonien non perturbé est hermitien). Cela conduit à la modification énergétique du premier ordre :
. Le premier terme de gauche s'annule avec le premier terme de droite (le hamiltonien non perturbé est hermitien). Cela conduit à la modification énergétique du premier ordre :C'est tout simplement l'espérance du hamiltonien de perturbation lorsque le système est dans l'état non perturbé. Ce résultat peut être interprété de la manière suivante : supposons qu'une perturbation soit appliquée, mais que l'on conserve le système dans l'état quantique
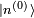 , qui est un état quantique valide bien que ne correspondant plus à un état propre de l'énergie. La perturbation fait que l'énergie moyenne de cet état croît de
, qui est un état quantique valide bien que ne correspondant plus à un état propre de l'énergie. La perturbation fait que l'énergie moyenne de cet état croît de 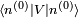 . Cependant la modification réelle de l'énergie est légèrement différente, car l'état propre perturbé n'est pas exactement
. Cependant la modification réelle de l'énergie est légèrement différente, car l'état propre perturbé n'est pas exactement 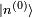 . Les modifications qui s'ensuivent sont données par les corrections des deuxième ordre et suivants de l'énergie.
. Les modifications qui s'ensuivent sont données par les corrections des deuxième ordre et suivants de l'énergie.
Avant de calculer les corrections à l'état propre d'énergie, on doit régler la question de la normalisation. On peut supposer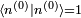 , mais la théorie de la perturbation postule que :
, mais la théorie de la perturbation postule que : 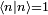 . Il s'ensuit qu'au premier ordre en , on doit avoir
. Il s'ensuit qu'au premier ordre en , on doit avoir 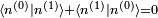 . Puisque la phase globale n'est pas déterminée en mécanique quantique, on peut postuler sans perte de généralité que
. Puisque la phase globale n'est pas déterminée en mécanique quantique, on peut postuler sans perte de généralité que 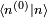 est réel. Ainsi,
est réel. Ainsi, 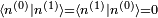 , et on en déduit :
, et on en déduit :Afin d'obtenir la correction
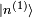 du premier ordre à l'état propre d'énergie, on utilise sa valeur extraite de l'équation au premier ordre ci-dessus. On utilise ensuite l'identité
du premier ordre à l'état propre d'énergie, on utilise sa valeur extraite de l'équation au premier ordre ci-dessus. On utilise ensuite l'identitéoù les
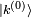 sont les états propres de situés dans le complément orthogonal de
sont les états propres de situés dans le complément orthogonal de 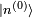 . Les termes proportionnels à se compensent, et le résultat est :
. Les termes proportionnels à se compensent, et le résultat est :Supposons pour le moment que le niveau d'énergie d'ordre zéro ne soit pas dégénéré, c'est-à-dire qu'il n'y ait pas d'état propre de
dans le complément orthogonal de d'énergie 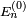 . On multiplie alors par
. On multiplie alors par 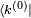 , ce qui donne :
, ce qui donne :et par conséquent le composant de la correction de premier ordre selon
par le postulat 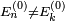 . Nous avons au total :
. Nous avons au total :La modification de premier ordre dans le
 -ième ket propre de l'énergie possède une contribution des états propres de l'énergie
-ième ket propre de l'énergie possède une contribution des états propres de l'énergie 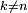 . Chaque terme est proportionnel à l'élément de matrice
. Chaque terme est proportionnel à l'élément de matrice 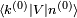 , qui est une mesure de combien la perturbation mélange l'état propre avec l'état propre
, qui est une mesure de combien la perturbation mélange l'état propre avec l'état propre  ; il est également inversement proportionnel à la différence d'énergie entre les états propres et , ce qui signifie que la perturbation « déforme » l'état propre plus facilement vers des états propres d'énergies voisines. On voit aussi que l'expression est singulière si l'un de ces états possède la même énergie que l'état , qui est ce pourquoi l'on postule la non-dégénérescence.
; il est également inversement proportionnel à la différence d'énergie entre les états propres et , ce qui signifie que la perturbation « déforme » l'état propre plus facilement vers des états propres d'énergies voisines. On voit aussi que l'expression est singulière si l'un de ces états possède la même énergie que l'état , qui est ce pourquoi l'on postule la non-dégénérescence.Corrections du deuxième ordre et suivants
On peut trouver les déviations d'ordres supérieurs par une méthode similaire, bien que les calculs deviennent plus compliqués avec la formulation employée. La condition de normalisation indique que :
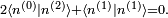 Jusqu'au deuxième ordre, les expressions pour les énergies et les états propres (normalisés) sont :
Jusqu'au deuxième ordre, les expressions pour les énergies et les états propres (normalisés) sont :En étendant la méthode précédente, on démontre que la correction d'énergie du troisième ordre est[3]
Effets de la dégénérescence
Supposons que deux ou plus états propres d'énergie sont dégénérés à l'énergie
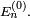 Les calculs précédents pour les modifications d'énergie du premier ordre ne sont pas affectés, mais le calcul de la modification de l'état propre est incorrect car l'opérateur
Les calculs précédents pour les modifications d'énergie du premier ordre ne sont pas affectés, mais le calcul de la modification de l'état propre est incorrect car l'opérateurn'a pas d'inverse dans le complément orthogonal de
.
Cela relève d'un problème conceptuel plutôt que mathématique. Imaginons que l'on ait deux ou plus états propres perturbés avec différentes énergies, continûment générés à partir d'un nombre égal d'états propres non perturbés dégénérés. Soit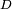 le sous-espace sous-tendu par ces états propres dégénérés, qui en forment donc une base particulière. Le problème repose sur le fait que tant que le système n'est pas perturbé, n'y a pas de méthode a priori pour choisir une base d'états propres de l'énergie. En choisissant comme base différentes combinaisons linéaires des états propres utilisés pour construire , les vecteurs de base ne génèreraient pas continûment les états propres perturbés.
le sous-espace sous-tendu par ces états propres dégénérés, qui en forment donc une base particulière. Le problème repose sur le fait que tant que le système n'est pas perturbé, n'y a pas de méthode a priori pour choisir une base d'états propres de l'énergie. En choisissant comme base différentes combinaisons linéaires des états propres utilisés pour construire , les vecteurs de base ne génèreraient pas continûment les états propres perturbés.On voit ainsi qu'en présence d'une dégénérescence, la théorie de la perturbation ne fonctionne pas avec un choix de base arbitraire. On doit plutôt choisir une base telle que le hamiltonien de perturbation soit diagonal dans le sous-espace dégénéré
. En d'autres termes,Dans ce cas, l'équation pour la perturbation de premier ordre dans l'état propre d'énergie se réduit à :
L'opérateur de gauche n'est pas singulier lorsqu'il est appliqué aux états propres n'appartenant pas à
, et on peut alors écrireThéorie de la perturbation dépendante du temps
La théorie de la perturbation dépendante du temps, développée par Paul Dirac, traite de l'effet d'une perturbation
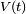 dépendante du temps appliquée à un hamiltonien indépendant du temps. Le hamiltonien perturbé étant dépendant du temps, ses niveaux et états propres d'énergie le sont aussi, et de toute manière leur interprétation est sujette à caution en vertu du principe d'incertitude, qui interdit de donner un sens physique à un instant donné à une énergie donnée. Par conséquent, les objectifs de la théorie de la perturbation dépendante du temps sont légèrement différents de ceux la théorie de la perturbation indépendante du temps. On traite les quantités suivantes :
dépendante du temps appliquée à un hamiltonien indépendant du temps. Le hamiltonien perturbé étant dépendant du temps, ses niveaux et états propres d'énergie le sont aussi, et de toute manière leur interprétation est sujette à caution en vertu du principe d'incertitude, qui interdit de donner un sens physique à un instant donné à une énergie donnée. Par conséquent, les objectifs de la théorie de la perturbation dépendante du temps sont légèrement différents de ceux la théorie de la perturbation indépendante du temps. On traite les quantités suivantes :- l'espérance mathématique dépendante du temps d'une observable
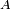 , pour un état initial donné.
, pour un état initial donné. - les amplitudes dépendantes du temps des états quantiques dans la base des kets propres (vecteurs propres) de l'énergie dans le système non perturbé.
La première quantité est importante car elle est à l'origine du résultat classique d'une mesure de
réalisée sur un nombre macroscopique d'exemplaires du système perturbé. Par exemple, on peut prendre comme le déplacement dans la direction  de l'électron dans un atome d'hydrogène, dans le cas duquel l'espérance mathématique, lorsqu'elle est multipliée par un coefficient approprié, donne la polarisation électrique dépendante du temps du gaz d'hydrogène. Avec un choix approprié de perturbation (comme par exemple un potentiel électrique oscillant), cela permet de calculer la permittivité diélectrique du gaz.
de l'électron dans un atome d'hydrogène, dans le cas duquel l'espérance mathématique, lorsqu'elle est multipliée par un coefficient approprié, donne la polarisation électrique dépendante du temps du gaz d'hydrogène. Avec un choix approprié de perturbation (comme par exemple un potentiel électrique oscillant), cela permet de calculer la permittivité diélectrique du gaz.
La seconde concerne la probabilité temporelle d'occupation de chaque état propre. Cela est particulièrement utile en physique des lasers, dans laquelle on s'intéresse à des populations dans différents états atomiques dans un gaz lorsqu'un champ électrique variable dans le temps est appliqué. Ces probabilités sont aussi utiles pour calculer l'élargissement quantique des raies spectrales.On exposera brièvement ci-après les idées de la formulation de Dirac de la théorie de la perturbation dépendante du temps. Choisissons une base d'énergie
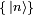 pour le système non perturbé. On ne portera plus les exposants pour les états propres, car parler de niveaux d'énergie et d'états propres pour le système perturbé est peu significatif.
pour le système non perturbé. On ne portera plus les exposants pour les états propres, car parler de niveaux d'énergie et d'états propres pour le système perturbé est peu significatif.Si le système non perturbé est un état propre
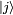 au temps
au temps 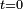 , son état aux temps suivants varie seulement d'une phase (on se place dans la représentation de Schrödinger, où les vecteurs d'état évoluent dans le temps et les opérateurs restent constants) :
, son état aux temps suivants varie seulement d'une phase (on se place dans la représentation de Schrödinger, où les vecteurs d'état évoluent dans le temps et les opérateurs restent constants) :On introduit alors une perturbation du hamiltonien, dépendante du temps
. Le hamiltonien du système perturbé est :Soit
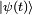 la notation pour l'état quantique du système perturbé au temps
la notation pour l'état quantique du système perturbé au temps  . Il obéit à l'équation de Schrödinger dépendante du temps,
. Il obéit à l'équation de Schrödinger dépendante du temps,L'état quantique à chaque instant peut être exprimé comme une combinaison linéaire de la base propre
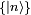 . On peut écrire la combinaison linéaire comme étant :
. On peut écrire la combinaison linéaire comme étant :où les
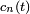 sont des fonctions complexes non déterminées de que nous appellerons amplitudes (à strictement parler, ce sont les amplitudes dans la représentation de Dirac). On a explicitement extrait les facteurs de phase exponentiels
sont des fonctions complexes non déterminées de que nous appellerons amplitudes (à strictement parler, ce sont les amplitudes dans la représentation de Dirac). On a explicitement extrait les facteurs de phase exponentiels 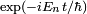 du côté droit de l'équation. C'est simplement un problème de convention, et peut être fait sans perte de généralité. La raison pour laquelle on s'intéresse à ce problème est que quand le système débute dans l'état et qu'il n'y a pas de perturbation, les amplitudes ont la propriété intéressante que, pour tout ,
du côté droit de l'équation. C'est simplement un problème de convention, et peut être fait sans perte de généralité. La raison pour laquelle on s'intéresse à ce problème est que quand le système débute dans l'état et qu'il n'y a pas de perturbation, les amplitudes ont la propriété intéressante que, pour tout , 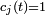 et
et 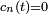 si
si 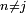 .
.Le carré de la valeur absolue de l'amplitude
est la probabilité que le système soit dans l'état au temps :En insérant l'expression de
dans l'équation de Schrödinger, et en calculant 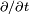 par dérivation des fonctions composées, on obtient :
par dérivation des fonctions composées, on obtient :En multipliant à gauche cette équation par
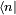 on réduit cette équation en un système d'équations différentielles couplées pour les amplitudes :
on réduit cette équation en un système d'équations différentielles couplées pour les amplitudes :Les éléments de matrice de
jouent un rôle similaire à celui tenu dans la théorie de la perturbation indépendante du temps, étant proportionnels au taux auquel les amplitudes sont modifiées entre les états. Il faut noter, cependant, que la direction (complexe) de la modification est conditionnée par le facteur de phase exponentiel. Sur des temps bien plus importants que 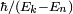 , la phase peut cycler plusieurs fois. Si la dépendance en temps de est suffisamment lente, cela peut provoquer une oscillation des amplitudes d'état. De telles oscillations sont utilisées pour gérer les transitions radiatives dans les lasers.
, la phase peut cycler plusieurs fois. Si la dépendance en temps de est suffisamment lente, cela peut provoquer une oscillation des amplitudes d'état. De telles oscillations sont utilisées pour gérer les transitions radiatives dans les lasers.
Jusque là, on n'a fait aucune approximation, donc l'ensemble d'équations différentielles est exact. En indiquant des valeurs initiales appropriées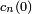 , on peut en principe trouver une solution exacte (non perturbative). Cela est facilement trouvé lorsqu'il y a seulement deux niveaux d'énergie (
, on peut en principe trouver une solution exacte (non perturbative). Cela est facilement trouvé lorsqu'il y a seulement deux niveaux d'énergie (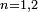 ), et la solution est utile pour des systèmes modèles comme la molécule d'ammoniac, ou une particule de spin
), et la solution est utile pour des systèmes modèles comme la molécule d'ammoniac, ou une particule de spin 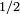 . Cependant, il est difficile de trouver des solutions exactes lorsqu'il y a plusieurs niveaux d'énergie, et l'on cherchera plutôt des solutions perturbatives, qui peuvent être obtenues en mettant les équations sous une forme intégrale :
. Cependant, il est difficile de trouver des solutions exactes lorsqu'il y a plusieurs niveaux d'énergie, et l'on cherchera plutôt des solutions perturbatives, qui peuvent être obtenues en mettant les équations sous une forme intégrale :On effectuant de manière répétée la substitution pour chaque
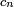 du côté droit, on obtient la solution itérative :
du côté droit, on obtient la solution itérative :où, par exemple, le terme de premier ordre est :
De nombreux résultats induits peuvent être obtenus, comme la règle d'or de Fermi, qui lie le taux de transition entre états quantiques à la densité d'états à énergies particulières, et les séries de Dyson, obtenues en appliquant la méthode itérative à l'opérateur d'évolution temporelle, qui est l'un des points de départ de la méthode des diagrammes de Feynman. La Théorie de la perturbation de Møller-Plesset est une application de la théorie de la perturbation à la méthode Hartree-Fock.
Voir aussi
- règle d'or de Fermi
- théorie des perturbations
Références
Bibliographie
- Albert Messiah, Mécanique quantique [détail des éditions], chapitre 16
- C. Cohen-Tannoudji, B. Diu et F. Laloë, Mécanique quantique [détail des éditions], chapitre 11
- (en) Cet article est partiellement ou en totalité issu d’une traduction de l’article de Wikipédia en anglais intitulé « Perturbation theory (quantum mechanics) ».
 Portail de la chimie
Portail de la chimie Portail de la physique
Portail de la physique
Catégories : Mécanique quantique | Chimie quantique
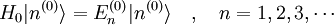
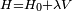
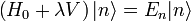
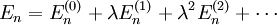
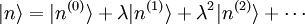
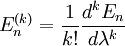
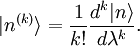
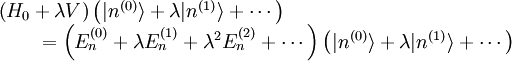
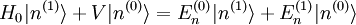
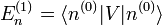
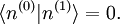
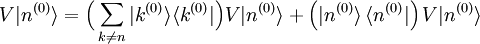
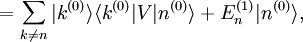
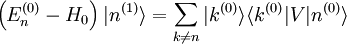
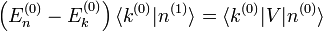
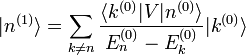
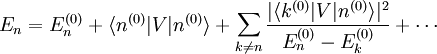
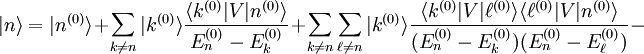
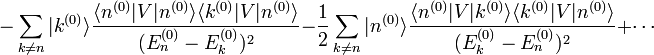
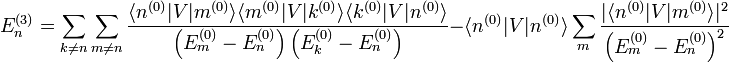
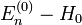
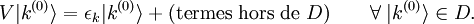
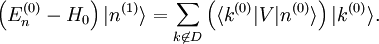
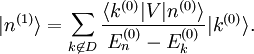
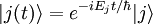
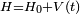
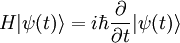
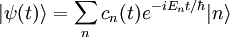
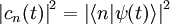
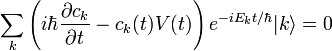
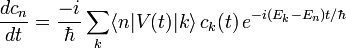
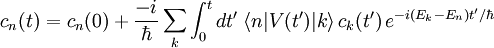
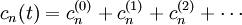
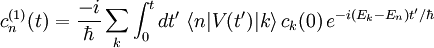
Wikimedia Foundation. 2010.