- Théorie de la fonctionnelle de la densité
-
 Pour les articles homonymes, voir DFT.
Pour les articles homonymes, voir DFT.La théorie de la fonctionnelle de la densité (pour Density Functional Theory, sous-entendu électronique : DFT) constitue au début du XXIe siècle l'une des méthodes les plus utilisées dans les calculs quantiques de la structure électronique de la matière (atomes, molécules, solides) aussi bien en physique de la matière condensée qu'en chimie quantique. La DFT trouve ses origines dans le modèle développé par Llewellyn Thomas et Enrico Fermi à la fin des années 1920. Néanmoins il faudra attendre le milieu des années 1960 et les contributions de Pierre Hohenberg, Walter Kohn et Lu Sham pour que soit établi le formalisme théorique sur lequel repose la méthode actuelle.
Les méthodes traditionnelles dans les théories de la structure électronique de la matière, en particulier la théorie Hartree-Fock et les méthodes dérivées de ce formalisme, se fondent sur une fonction d'onde multiélectronique. L'objectif principal de la théorie de la fonctionnelle de la densité est de remplacer la fonction d'onde multiélectronique par la densité électronique en tant que quantité de base pour les calculs. Alors que la fonction d'onde multiélectronique dépend de 3N variables (où N est le nombre total de particules du système), la densité est seulement fonction de trois variables ; il s'agit donc d'une quantité plus facile à traiter tant mathématiquement que conceptuellement. Le principe de la DFT consiste en une reformulation du problème quantique à N corps en un problème monocorps (ou, à la rigueur, bi-corps si l'on considère les problèmes de spin) avec pour paramètre la densité électronique. L'idée centrale de la DFT est que la seule densité électronique de l'état fondamental du système détermine entièrement les valeurs moyennes des observables, comme par exemple l'énergie.
La DFT a été à l'origine principalement développée dans le cadre de la théorie quantique non-relativiste (équation de Schrödinger indépendante du temps) et dans l'approximation de Born-Oppenheimer. La théorie fut par la suite étendue au domaine de la mécanique quantique dépendante du temps (on parle alors de TDDFT pour Time-Dependent Density Functional Theory) et au domaine relativiste. La DFT est également utilisée pour la description thermodynamique des fluides classiques.
Sommaire
Notions de base
« Les lois physiques fondamentales nécessaires à la théorie mathématique d'une grande partie de la physique et de la totalité de la chimie sont ainsi complètement connues, et la difficulté est seulement que l'application exacte de ces lois mène à des équations beaucoup trop complexes pour être résolues »
— Paul A.M. Dirac, 1929[1]
Equation de Schrödinger
Article détaillé : équation de Schrödinger.L'équation fondamentale à résoudre pour décrire la structure électronique d'un système à plusieurs noyaux et électrons est l'équation établie par Erwin Schrödinger (1887-1961) en 1925[2], appelée depuis équation de Schrödinger, et qui s'écrit[3] :
![H\Psi = \left[- \sum_i^N \frac{\hbar^2}{2m}\nabla_i^2 - \sum_{I}^A \frac{\hbar^2}{2M}\nabla_I^2 - \sum_{i,I} \frac{Z_I e^2}{|\vec r_i - \vec R_I|} + \sum_{i<j} \frac{e^2}{|\vec r_i - \vec r_j|} + \sum_{I<J} \frac{Z_I Z_J e^2}{|\vec R_I - \vec R_J|}\right] \Psi= E\Psi](e/00ec9266004feb19eb80fd7f2f6c2ccf.png)
où
 est l'hamiltonien moléculaire et
est l'hamiltonien moléculaire et  la fonction d'onde. Les deux premiers termes de l'hamiltonien sont respectivement les opérateurs énergie cinétique des
la fonction d'onde. Les deux premiers termes de l'hamiltonien sont respectivement les opérateurs énergie cinétique des  électrons (indexés
électrons (indexés  ) et des
) et des  noyaux atomiques (indexés
noyaux atomiques (indexés  ). Les trois autres termes représentent les différents potentiels d'interaction électron-noyau, électron-électron et noyau-noyau.
). Les trois autres termes représentent les différents potentiels d'interaction électron-noyau, électron-électron et noyau-noyau.Sous cette forme, l'équation de Schrödinger est trop complexe pour pouvoir être résolue analytiquement. De manière à simplifier la résolution de cette équation, Max Born (1882-1970) et Robert Oppenheimer (1904-1967) ont proposé une approximation visant à simplifier l'équation de Schrödinger[4]. L'approximation de Born-Oppenheimer considère la position des noyaux atomiques comme fixes ; leur énergie cinétique peut donc être négligée et le terme d'interaction entre noyaux considéré comme une constante (que l'on notera
 ). Cette approximation se justifie par le rapport de masse entre les particules constitutives du noyau (protons et neutrons) et les électrons[5]. L'équation à résoudre s'écrit alors :
). Cette approximation se justifie par le rapport de masse entre les particules constitutives du noyau (protons et neutrons) et les électrons[5]. L'équation à résoudre s'écrit alors :![H\Psi = \left[- \sum_i^N \frac{\hbar^2}{2m}\nabla_i^2 - \sum_{i,I} \frac{Z_I e^2}{|\vec r_i - \vec R_I|} + \sum_{i<j} \frac{e^2}{|\vec r_i - \vec r_j|} + E_{II}\right] \Psi= E\Psi](a/44a3f2fac71f6a76964e8ab9835f5276.png)
De manière à alléger les notations, on représentera, par convention, l'opérateur énergie cinétique par
 , le potentiel externe ressenti par les électrons par
, le potentiel externe ressenti par les électrons par  et le potentiel d'interaction électron-électron par
et le potentiel d'interaction électron-électron par  . L'équation s'écrit dès lors sous une forme plus condensée comme[6] :
. L'équation s'écrit dès lors sous une forme plus condensée comme[6] :![\,\! H\Psi = [T + V_{ext} + U]\Psi = E\Psi](6/fb608fc81b50578bf864736cee1bbd1d.png)
De nombreuses méthodes ont été développées pour résoudre l'équation de Schrödinger multiélectronique en décrivant par exemple la fonction d'onde comme un déterminant de Slater ; c'est le cas de la méthode Hartree-Fock. La DFT fournit une méthode alternative en considérant comme quantité de base pour la description du système la densité électronique.
Densité électronique
Définition et propriétés
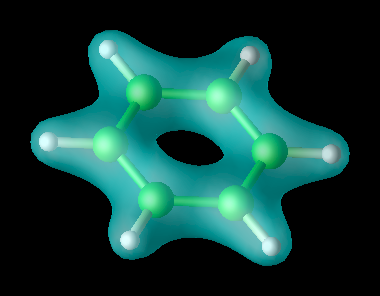
 Représentation de la densité électronique pour une molécule de benzène dans son état fondamental.
Représentation de la densité électronique pour une molécule de benzène dans son état fondamental.
La probabilité de trouver un électron parmi les
électrons du système dans un élément de volume  centré sur la position
centré sur la position  s'exprime comme :
s'exprime comme :
où
 est la densité de probabilité électronique qui est définie comme[7] :
est la densité de probabilité électronique qui est définie comme[7] :
La densité de probabilité possède notamment deux propriétés importantes :
Densité de paire
La densité de paire est la probabilité de trouver simultanément 2 des N électrons du système dans les éléments de volume
et  et est notée
et est notée 

Modèle de Thomas-Fermi
La Théorie de la Fonctionnelle de la Densité tire ses origines du modèle de Thomas-Fermi , développé par énergie cinétique, représentée comme une fonctionnelle de la densité électronique :
![T_{TF}[n]=\frac{3}{10}(3\pi^2)^{2/3}\int n^{5/3}(r) d^3r.](5/fd5926f8552fdc84425a8ba70a05d01b.png)
De cette manière, il leur a été possible de calculer l’énergie d’un atome, en utilisant cette fonctionnelle d’énergie cinétique combinée avec l’expression classique des interactions noyau–électron et électron–électron qui peuvent eux aussi être exprimées en termes de densité électronique.
![E_{TF}[n]=\frac{3}{10}(3\pi^2)^{2/3}\int n^{5/3}(r) d^3r + \int{ V_{ext}(r)n(r)dr} + \frac{1}{2} \int{\frac{n(r)n(r')}{|r-r'|}drdr'}](0/c800a2c9c1f7f97a783e9c0010d02f5b.png)
Bien que cela soit une importante première étape, la précision de l’équation de Thomas-Fermi reste cependant limitée, parce que la fonctionnelle de l’énergie cinétique résultante est approximée mais aussi parce que cette méthode ne tient pas compte de l’énergie d’échange d’un atome, conséquence du principe de Pauli, ni de la corrélation électronique. Une fonctionnelle d’échange énergétique fut ajoutée par Paul Dirac (1902-1984) en 1930[12].
Cependant, la méthode de Thomas-Fermi-Dirac reste relativement imprécise pour la plupart des applications, la plus grande source d’erreur provenant de l’écriture de l’énergie cinétique qui peut toutefois être améliorée en y ajoutant la correction proposée en 1935 par Carl von Weizsäcker (1912-2007) qui prend en compte le gradient de la densité dans l'expression de la fonctionnelle de l'énergie cinétique[13] :
![T_W[n]=\frac{1}{8}\frac{\hbar^2}{m}\int\frac{|\nabla n(r)|^2}{n(r)}dr](5/635a79018ee06cd0c59802c7b933ad76.png)
La methode de Thomas-Fermi a été notamment utilisée pour les équations d'états des éléments chimiques[14] mais sa portée ne peut être guère plus étendue. Edward Teller (1908-2003) a en effet montré en 1962 que la théorie de Thomas-Fermi était incapable de decrire la liaison moléculaire[15].
Formalisme mathématique
Théorèmes de Hohenberg et Kohn
L'approche développée par Pierre Hohenberg et Walter Kohn est de reformuler la théorie de la fonctionnelle de la densité proposée par Thomas et Fermi par une théorie exacte d'un système à plusieurs corps. La formulation est applicable pour tout système de particules en interaction évoluant dans un potentiel externe et repose sur deux théorèmes essentiels qui furent énoncés et démontré par Hohenberg et Kohn dans leur article de 1964[16].
Premier Théorème : Pour tout système de particules en interaction dans un potentiel externe Vext(r), le potentiel Vext(r) est uniquement déterminé, à une constante additive près, par la densité n0(r) de la particule dans son état fondamental. Preuve du Théorème ILe Premier Théorème HK peut se démontrer très simplement en utilisant un raisonnement par l'absurde. Supposons qu'il puisse exister deux potentiels externes différents
 et
et  (par différent, on entend autrement que par une constante additive) associés à la densité de l'état fondamental . Ces deux potentiels vont conduire à deux hamiltoniens différents
(par différent, on entend autrement que par une constante additive) associés à la densité de l'état fondamental . Ces deux potentiels vont conduire à deux hamiltoniens différents  et
et  dont les fonctions d'ondes
dont les fonctions d'ondes  et
et  décrivant l'état fondamental sont différentes. Comme ne décrit par l'état fondamental de on peut donc écrire que :
décrivant l'état fondamental sont différentes. Comme ne décrit par l'état fondamental de on peut donc écrire que :
Cette inégalité stricte est valable si l'état fondamental est non dégénéré ce qui est supposé dans le cas de l'approche de HK. Le dernier terme de l'expression précédente peut s'écrire :

![= E^{(2)}+ \int [V_{ext}^1(r)-V_{ext}^2(r)]n_0(r)d^3r](a/18a84239a9168abcf6dc3a12f3291303.png)
ce qui donne :
![E^{(1)}< E^{(2)}+ \int [V_{ext}^1(r)-V_{ext}^2(r)]n_0(r)d^3r](c/18ccd21ce6437563ae25f7cbc85f4908.png)
Il va également de soi que le même raisonnement peut être réalisé en considérant
 à la place de
à la place de  . On obtient alors la même équation que précédemment, les symboles (1) et (2) étant inversés :
. On obtient alors la même équation que précédemment, les symboles (1) et (2) étant inversés :![E^{(2)}< E^{(1)}+ \int [V_{ext}^2(r)-V_{ext}^1(r)]n_0(r)d^3r](5/ff5375f459594e68ea6208a4566c8371.png)
On additionnant membre à membre ces deux dernières équations, on obtient l'égalité contradictoire suivante :

Le schéma ci-dessous illustre l'apport que fournit le premier théorème de Hohenberg et Khom. Les simples flèches indiquent le schéma obtenu dans le cas de la résolution traditionnelle de l'équation de Schrödinger où la connaissance du potentiel externe va permettre de déterminer les différents états électroniques (c'est-à-dire les fonctions d'ondes) ainsi que l'état fondamental et la densité électronique qui lui est associée. Le premier théorème HK, représenté par la double flèche, permet de compléter ce cycle. Cela signifie que toutes les propriétés du système peuvent être complètement déterminées si l'on connait la densité électronique de l'état fondamental.
![\begin{array}[t]{lcl} V_{ext}(r) &\Longleftarrow & n_0(r) \\
\downarrow & & \uparrow \\
\Psi_i({r}) & \rightarrow & \Psi_0({r})
\end{array}](c/9ec96542bbe6e1c85d2140cc128be5de.png)
Second Théorème : Il existe une fonctionnelle universelle E[n] exprimant l'énergie en fonction de la densité électronique n(r), valide pour tout potentiel externe Vext(r). Pour chaque Vext(r) particulier, l'énergie de l'état fondamental du système est la valeur qui minimise cette fonctionnelle, la densité n(r) qui lui est associée correspond à la densité exacte n0(r) de l'état fondamental. Preuve du Théorème IIToutes les propriétés du système étant définies par la connaissance de la densité
 , elles peuvent donc être décrites comme des fonctionnelles de la densité. L'énergie totale peut donc s'écrire :
, elles peuvent donc être décrites comme des fonctionnelles de la densité. L'énergie totale peut donc s'écrire :![E_{HK}[n]=T[n]+ U[n]+ \int V_{ext}(\vec r)n(\vec r) d^3r + E_{II}](2/6a2487efb62dcfbe26afb39444e0d56a.png)
![= F_{HK}[n]+\int V_{ext}(\vec r)n(\vec r) d^3r + E_{II}](2/eb2148ce72015c02204bcdac720c4d0b.png)
où
exprime l'énergie d'interaction entre les noyaux atomiques tandis que la fonctionnelle ![\,\! F_{HK}[n]](d/c5df61846ac602e226d6a35e8fbbcea6.png) inclut toutes les contributions électroniques (énergies cinétique et potentielle). Cette fonctionnelle étant la même pour tout système électronique (et indépendante du potentiel externe), elle est appelée fonctionnelle universelle.
inclut toutes les contributions électroniques (énergies cinétique et potentielle). Cette fonctionnelle étant la même pour tout système électronique (et indépendante du potentiel externe), elle est appelée fonctionnelle universelle.![\,\! F_{HK}[n] = T[n]+ U[n]](0/750787b0889c3789eb147cad92dfc02a.png)
Considérons à présent un système ayant une densité à l'état fondamental noté
 et qui correspond à un potentiel externe
et qui correspond à un potentiel externe  . La fonctionnelle de Hohenberg et Kohn est égale à la valeur obtenue pour l'hamiltonien de l'état fondamental associé à la fonction d'onde
. La fonctionnelle de Hohenberg et Kohn est égale à la valeur obtenue pour l'hamiltonien de l'état fondamental associé à la fonction d'onde  :
:![E^{(1)} = F_{HK}[n^{(1)}] = \langle \Psi^{(1)}|H^{(1)}|\Psi^{(1)}\rangle](1/ad18071706508e1530e89a468ece548c.png)
Si nous considérons à présent une densité différente
 qui correspond nécessairement à une fonction d'onde
qui correspond nécessairement à une fonction d'onde  différente de . On constate de manière immédiate que l'énergie
différente de . On constate de manière immédiate que l'énergie  caractérisant cet état est plus élevée que (l'état 1 est l'état fondamental et est unique par hypothèse).
caractérisant cet état est plus élevée que (l'état 1 est l'état fondamental et est unique par hypothèse).
 est forcément moindre que la valeur obtenue avec n'importe quelle autre densité
est forcément moindre que la valeur obtenue avec n'importe quelle autre densité  . De ce fait on peut en déduire que, si la fonctionnelle universelle est connue, la minimisation de l'énergie totale par une variation de la fonction densité va nous conduire à la densité (et donc l'énergie) de l'état fondamental.
. De ce fait on peut en déduire que, si la fonctionnelle universelle est connue, la minimisation de l'énergie totale par une variation de la fonction densité va nous conduire à la densité (et donc l'énergie) de l'état fondamental.Le second théorème montre que l'énergie apparaît comme une fonctionnelle de la densité, et que pour tout potentiel extérieur, la densité qui minimise cette fonctionnelle est la densité exacte de l'état fondamental.
En conclusion, les deux théorèmes proposés par Hohenberg et Kohn permettent de déplacer le problème posé par la résolution d’une équation de Schrödinger multiélectronique. En effet, la méthode DFT nous enseigne que si la forme de la fonctionnelle est connue, il est relativement aisé, pour un potentiel externe donné, de déterminer l’énergie de l'état fondamental. Le problème qui se pose est alors la formulation de la fonctionnelle
![\,\! F[n]](c/84c035108cf511edb68e07f4b3c0c829.png) et en particulier l’expression de l'énergie cinétique
et en particulier l’expression de l'énergie cinétique ![\,\! T[n]](2/2220bca035dc1ca4b3c43a0465cf7de4.png) . En effet, il n’est pas possible, pour un système de N électrons en interaction, de trouver une expression analytique à la fonctionnelle de l’énergie cinétique.
. En effet, il n’est pas possible, pour un système de N électrons en interaction, de trouver une expression analytique à la fonctionnelle de l’énergie cinétique.Ansatz de Kohn et Sham
« Si vous n'aimez pas la réponse, modifiez la question. »
— [17]
 Schéma décrivant le processus itératif pour la résolution des équations de Kohn-Sham
Schéma décrivant le processus itératif pour la résolution des équations de Kohn-ShamL’énergie cinétique d’un gaz d’électrons en interaction étant inconnue, Walter Kohn (1923-) et Lu Sham ont proposé en 1965 un ansatz qui consiste à remplacer le système d'électrons en interaction, impossible à résoudre analytiquement, par un problème d'électrons indépendants évoluant dans un potentiel externe[18].
Mathématiquement, cela revient à exprimer la fonctionnelle énergie totale de Hohenberg et Kohn décrite comme :
![E_{HK}[n] = F[n] + \int V(r)n(r)dr](2/472cb33d0bdcd630ae43055aaaf3c7ce.png)
par l'expression suivante :
![\,\! E_{S}[n] = T_{S}[n] + V_{S}](7/9578633f5d77d53caeaf7dbc2b736670.png)
où
![\,\! T_{S}[n]](4/f147bbb38e56b65ba9f257d99d510a2c.png) est l'énergie cinétique des électrons sans interaction et
est l'énergie cinétique des électrons sans interaction et ![\,\! V_{S}[n]](2/832fa6ccda8cdadf2eadcd71a1a10de1.png) le potentiel dans lequel les électrons se déplacent. La densité électronique
le potentiel dans lequel les électrons se déplacent. La densité électronique  est strictement égale à la densité apparaissant dans la fonctionnelle définie par Hohenberg et Kohn si le potentiel externe est défini comme :
est strictement égale à la densité apparaissant dans la fonctionnelle définie par Hohenberg et Kohn si le potentiel externe est défini comme :
C'est-à-dire si celui-ci inclut la correction à l'énergie cinétique suite à l'ansatz de Kohn et Sham. L'intérêt de la reformulation introduite par Kohn et Sham est que l'on peut maintenant définir un hamiltonien monoélectronique et écrire les équations de Kohn-Sham monoélectroniques qui, contrairement à l'équation de Schrödinger définie plus haut, peuvent être résolues analytiquement.
![\left[-\frac{\hbar^2}{2m}\nabla^2+V_s(\vec r)\right] \phi_i(\vec r) = \epsilon_i \phi_i(\vec r),](4/964453454685b61c5d473369e8e21610.png)
La résolution des équations de Kohn-Sham va permettre de déterminer les orbitales
 qui vont reproduire la densité électronique du système multiélectronique d'origine.
qui vont reproduire la densité électronique du système multiélectronique d'origine.
Le potentiel effectif monoélectronique apparaissant dans l'équation peut être exprimé de manière plus détaillée comme :
![V_s = V + \int \frac{n_s(\vec r\,) n_s(\vec r\,')}{|\vec r-\vec r\,'|} {\rm d}^3r' + V_{\rm XC}[n_s(\vec r)]](9/fb9238fc18a1405d4e50111ccc4c624d.png)
Le premier terme est le potentiel externe créé par les noyaux, le deuxième exprime l'interaction coulombienne classique entre paire d'électrons (et est également appelé potentiel Hartree). Le dernier terme est le potentiel d'échange-corrélation et contient, outre l'échange et la corrélation électronique, les corrections à l'énergie cinétique. Celle-ci n'est pas connue exactement, le choix d'une fonction d'échange corrélation approximée constitue l'un des principaux choix d'approximation en DFT dans l'approche Kohn-Sham.
Comme on peut l'observer dans l'équation, ce potentiel dépend de la densité électronique, qui elle-même est calculée à partir des fonctions d'ondes des électrons indépendants, qui elle-même dépend du potentiel calculé à partir de la densité , etc. Cette approche conduit donc à un traitement dit self-consistent field (ou méthode du champ auto-cohérent) : en partant d'une valeur arbitraire de départ, on calcule en boucle les valeurs de densité, potentiel et fonctions d'ondes jusqu'à une situation stable où ces différentes valeurs n'évoluent presque plus.
Résolution numérique des équations de K-S
L'ansatz de Kohn et Sham permet d'aboutir à un ensemble d'équations de Schrödinger monoélectroniques connues sous le nom d'équations de Kohn-Sham :
![\Big[ - \frac{\nabla^2}{2m} + V_{ext} + V_H + V_{xc} \Big]\phi_i = \epsilon_i \phi_i](c/cdcae6cd76b92bd02b3bcfde7f4f76dc.png)
qui doivent être résolues numériquement selon un processus itératif. De manière à pouvoir résoudre ces équations de manière numérique, un certain nombre d'approximations peuvent ou doivent être envisagées. Klaus Capelle recense ainsi trois types d'approximations qui peuvent globalement être distinguées en DFT[19]. L'une est purement conceptuelle et concerne l'interprétation à donner aux valeurs propres
 obtenues après résolution[20]. Il ne s'agit donc pas exactement d'une approximation mais plutôt d'une réflexion sur la signification physique des valeurs propres. Le deuxième type d'approximation est d'ordre "technique" et concerne les choix effectués pour simplifier la résolution des équations ; il s'agit principalement du choix des fonctions de bases et de la réduction du nombre d'électrons à prendre en considération dans les calculs (c'est-à-dire l'utilisation de pseudopotentiel). Ces deux approches seront brièvement décrites ci-dessous.
obtenues après résolution[20]. Il ne s'agit donc pas exactement d'une approximation mais plutôt d'une réflexion sur la signification physique des valeurs propres. Le deuxième type d'approximation est d'ordre "technique" et concerne les choix effectués pour simplifier la résolution des équations ; il s'agit principalement du choix des fonctions de bases et de la réduction du nombre d'électrons à prendre en considération dans les calculs (c'est-à-dire l'utilisation de pseudopotentiel). Ces deux approches seront brièvement décrites ci-dessous.Choix des fonctions de base
Utilisation de pseudopotentiels
Article détaillé : Pseudo-potentiel.Approximations
Comme décrit plus haut la théorie DFT est, au stade des équations de Kohn-Sham, une théorie parfaitement exacte (mises à part l'approximation de Born-Oppenheimer et les approches numériques discutées précédemment) dans la mesure où la densité électronique qui minimise l'énergie totale est exactement la densité du système de N électrons en interaction. Cependant, la DFT reste inapplicable car le potentiel d'échange-corrélation (contenant également la correction à l'énergie cinétique) reste inconnu. Il est donc nécessaire d'approximer ce potentiel d'échange-corrélation. Deux types d'approximations existent : l'approximation de la densité locale ou LDA et l'approximation du gradient généralisé ou GGA ainsi que les méthodes dérivées qui se fondent sur une approche non locale.
LDA ou approximation de la densité locale
L'approche de la densité locale est fondée sur le modèle du gaz uniforme d'électron et constitue l'approche la plus simple pour exprimer l'énergie d'échange-corrélation. Celle-ci est décrite comme :
![E_{xc}[n] = \int n(\vec r)\varepsilon _{xc}[n]dr](9/c49ec3ed553208e6c0ae4d67d86e8907.png)
où εxc[n] désigne l'énergie d'échange-corrélation pour une particule d'un gaz homogène d'électron. La fonction εxc[n] peut être décomposée en une contribution d'échange εx[n] et de corrélation εc[n][21] :
εxc[n] = εx[n] + εc[n] La contribution provenant de l'échange électronique dans l'approximation de la densité locale est connue et provient de la fonctionnelle d'énergie d'échange formulée par Dirac[22]

L'approximation LDA peut être formulée de manière plus générale en prenant en compte le spin de l'électron dans l'expression de la fonctionnelle, on parle alors d'approximation LSDA (pour local spin density approximation). Cette approche fut initialement proposée par John C. Slater (1900-1976)[23] et permet de résoudre certains problèmes liés à une approche LDA, notamment le traitement de systèmes soumis à des champs magnétiques et les systèmes où les effets relativistes deviennent importants. En prenant en compte l'approximation LSDA, la fonctionnelle d'échange est exprimée comme :

où α et β expriment les spins up et down[24].
Pour l'énergie de corrélation, des valeurs précises sont disponibles via les calculs de Monte Carlo quantique établi par Ceperley[25]et par Ceperley et Alder[26] dont les résultats peuvent être interpolés afin d'obtenir une forme analytique. Il existe donc de nombreuses paramétrisations pour l'énergie de corrélation telles que, par exemple, celles de Hedin-Lundqvist [27], Perdew-Zunger[28] ou Volko-Wilkes-Nusair[29]
Forme mathématique de quelques potentiels de corrélationDans l'approximation LDA (ou LSDA), le potentiel de corrélation est défini par[30] :

où
 est l'énergie de corrélation et
est l'énergie de corrélation et  un paramètre qui décrit le rayon d'une sphère contenant en moyenne un électron dans un système électronique homogène de densité [31].
un paramètre qui décrit le rayon d'une sphère contenant en moyenne un électron dans un système électronique homogène de densité [31].
- Forme de Hedin-Lundqvist[32]
![\epsilon_c(r_s)^{HL} = - \frac{Ce^2}{2}\Big[(1+x^3)log(1+\frac{1}{x})+\frac{x}{2}-x^2-\frac{1}{3}\Big]](3/d43991d977e42029eea9b4c3e7064994.png)
où A = 21, C = 0.045 et x = rs/A
![V_c(r_s)^{HL} = - \frac{Ce^2}{2}log\Big[1+\frac{1}{x}\Big]](3/0c3bcc3a23be79da12d4b0711053eb99.png)
« L'approximation LSD est ainsi une approximation "first-principle", dans le sens où ses paramètres ne sont pas interpolés empiriquement à des résultats calculés ou expérimentaux autres que ceux pour lesquels sa forme est exacte. »
— J. Perdew et al (1996)[33]
Bien qu'étant une approche assez simple conceptuellement, l'approximation LDA permet néanmoins d'obtenir de bons résultats. Une compensation des erreurs permet d'expliquer en partie le relatif succès de la méthode LDA. Celle-ci tend en effet à sous-estimer l'énergie d'échange alors qu'elle surestime l'énergie de corrélation ce qui permet, in fine d'obtenir des valeurs assez bonnes pour l'énergie d'échange-corrélation.
GGA ou approximation des gradients généralisée
L'approche LDA se fondait sur le modèle du gaz d'électrons et supposait donc une densité électronique uniforme. Cependant les systèmes atomiques ou moléculaires sont le plus souvent très différents d'un gaz d'électrons homogène et, de manière plus générale, on peut considérer que tous les systèmes réels sont inhomogènes c'est-à-dire que la densité électronique possède une variation spatiale. Les méthodes dites GGA (Generalized gradient approximation), parfois aussi appelées méthodes non locales, ont été développées de manière à prendre en compte cette variation de la densité en exprimant les énergies d'échanges et de corrélation en fonction de la densité mais également de son gradient (c'est-à-dire sa dérivée première). De manière générale , l'énergie d'échange-corrélation est définie dans l'approximation GGA comme :
![E_{xc}^{GGA}[n_{\alpha}, n_{\beta}] = \int n(\vec r) \epsilon_{xc}[n_{\alpha}, n_{\beta}, \nabla n_{\alpha}, \nabla n_{\beta}] d^3r](f/5afef5d89272641956c8a1c4e2ecde03.png)
Globalement, les fonctionnelles GGA sont construites selon deux types de procédures différents. L'un est de nature empirique et consiste en une interpolation numérique des résultats expérimentaux obtenus sur un grand nombre de molécules. On peut citer comme exemple de fonctionnelle construite selon ce processus les fonctionnelles d'échange notée B (Becke88)[34], PW (Perdew-Wang)[35] ou bien encore mPW (modified Perdew-Wang)[36]. La deuxième procédure consiste à construire les fonctionnelles sur la base des principes de la mécanique quantique (et est en ce sens plus rationnelle). Les fonctionnelles d'échange B88 (Becke88)[37], P (Perdew86)[38] ou PBE (Perdew-Burke-Ernzerhof)[39] sont construites de cette manière.
Description mathématique de l'échange en GGADans l'approximation GGA, la fonctionnelle d'échange est exprimée comme[40] :
![E_x^{GGA}[n] = \int n(r) \epsilon_x^{hom.}[n(r)]f(\zeta)d^3r](f/87fc4b6c6c45239fb174211a9ab6ea6f.png)
où
 est l'énergie d'échange pour un gaz homogène d'électrons et
est l'énergie d'échange pour un gaz homogène d'électrons et  un facteur d'amélioration (en anglais enhancement factor) avec
un facteur d'amélioration (en anglais enhancement factor) avec  la variable sans dimension
la variable sans dimension![\zeta = \frac{|\nabla n|^2}{[2(3\pi^2)^{1/3}]^2 n^{5/3}}](6/be649c5328debde344c34bbcbebe7c69.png)
Au-delà de GGA, l'échelle de Jacob
Article principal : fonctionnelle hybride.Les méthodes GGA permettent d'obtenir une amélioration des résultats par rapport à une approche locale. Cependant, comme décrit plus haut, l'approche GGA n'est pas toujours suffisante pour une description correcte de diverses propriétés chimiques des composés. C'est pourquoi, à partir du milieu des années 1990, de nouveaux types de fonctionnelles ont été développées de manière à aller au-delà des résultats fournis par des méthodes GGA. Les fonctionnelles dites meta-GGA (ou m-GGA) font ainsi intervenir dans les équations le laplacien (c'est-à-dire la dérivée seconde) de la densité. Celles-ci permettent un gain de précision dans la détermination des propriétés moléculaires mais posent certains problèmes au niveau de la stabilité numérique. On peut citer comme exemple de fonctionnelle m-GGA, la fonctionnelle de corrélation B95 développée par Becke[41]. Un degré de précision supplémentaire est atteint en combinant l'échange et la corrélation obtenu par des méthodes GGA avec un certain pourcentage d'échange décrit par la théorie Hartree-Fock[42]. Les fonctionnelles construites sur ce principe sont qualifiées de fonctionnelles hybrides, on parle alors de fonctionnelles H-GGA (hybrid-GGA functional). La détermination du pourcentage d'échange Hartree-Fock à inclure dans la fonctionnelle est essentiellement déterminée de manière empirique. L'utilisation de ce type de fonctionnelle permet une amélioration significative des résultats et est devenue depuis plusieurs années le choix le plus populaire dans le domaine de la chimie quantique. La fonctionnelle d'échange-corrélation hybride B3LYP représentait ainsi 80% d'utilisation sur la période 1990-2006[43]. Les fonctionnelles HM-GGA (Hybrid-Meta GGA functional) représentent une nouvelle classe de fonctionnelles et font actuellement l'objet de nombreux développements. Le concept est similaire à l'approche des fonctionnelles hybrides, la différence est que l'on part de fonctionnelle m-GGA à la place de GGA. Ces fonctionnelles font donc intervenir l'échange Hartree-Fock, la densité électronique et son gradient ainsi que la densité électronique de l'énergie cinétique (c'est-à-dire le laplacien de la densité). C'est la cas, par exemple, de la fonctionnelle B1B95[41].
« [Jacob] eut un songe. Et voici, une échelle était appuyée sur la terre, et son sommet touchait au ciel. Et voici, les anges de Dieu montaient et descendaient par cette échelle. »
La métaphore de l'échelle de Jacob est due à J. Perdew[44] et illustre le progrès croissant dans le domaine des fonctionnelles de la densité depuis le milieu des années 1980. L'échelle de Jacob, telle que vue par Perdew, contient 5 échelons représentant les 5 générations de fonctionnelles. Les utilisateurs prennent ici la place des anges, montant ou descendant les barreaux selon ses besoins qui résultent d'un compromis entre exactitude et ressources informatiques disponibles.
 Die Engelsleiter (1691) du peintre Michael Willmann représentant la montée des anges le long de l'échelle de Jacob.
Die Engelsleiter (1691) du peintre Michael Willmann représentant la montée des anges le long de l'échelle de Jacob.Paradis = exactitude Échelon Méthode Exemple 5e échelon
Totalement non local
-
4e échelon
Hybrid Meta GGA
B1B95
Hybrid GGA
B3LYP
3e échelon
Meta GGA
BB95
2e échelon
GGA
BLYP
1e échelon
LDA
SPWL
Terre = Théorie Hartree-Fock
Nomenclature des fonctionnelles : l'exemple de B3LYP
Les fonctionnelles d'échange et de corrélation peuvent adopter des formes mathématiques souvent complexes. De manière à simplifier les notations, la convention est de noter les fonctionnelles du nom de leur(s) auteur(s) suivi de la date de publication dans le cas ou un même groupe a publié plusieurs fonctionnelles différentes. La fonctionnelle d'échange électronique développée par Axel Becke en 1988 est ainsi notée B et la fonctionnelle de corrélation publiée par le même auteur en 1995 est notée B95. Dans le cas où plusieurs auteurs sont impliqués dans le développement, les initiales de ceux-ci sont utilisées pour symboliser la fonctionnelle. La fonctionnelle de corrélation LYP est ainsi nommée du nom de ses trois auteurs Lee, Yang et Parr.
La description complète de l'échange et de la corrélation électronique est obtenue en combinant une fonctionnelle d'échange et une fonctionnelle de corrélation. La fonctionnelle est alors symbolisée en ajoutant simplement les symboles des fonctionnelles d'échange et de corrélation (toujours dans cet ordre). Dans les cas plus complexe des fonctionnelles hybrides le nombre de paramètres impliqués est également mentionné.
B3LYP est actuellement la fonctionnelle la plus employée en théorie DFT. Il s'agit d'une fonctionnelle hybride obtenue par combinaison linéaire entre des fonctionnelles d'échange et de corrélation GGA et de l'échange Hartree-Fock. B3LYP signifie Becke - 3 paramètres - Lee, Yang,Parr et est décrite comme :

Principales fonctionnelles d'échange-corrélation
Le tableau ci-dessous renseigne les principales fonctionnelles d'échange-corrélation employées dans les calculs DFT classées selon le type d'approximation utilisé. Une description plus complète ainsi que les références relatives aux diverses fonctionnelles reprises dans le tableau peuvent être trouvée dans le review publié par S.F. Sousa et al.[43]
Tableau des principales fonctionnelles d'échange-corrélationType Fonctionnelle Année Fonctionnelle d'échange Pourcentage d'échange HF Fonctionnelle de corrélation LSDA
SVWN3 1981 Slater 0 VWN n°3 SVWN5 1981 Slater 0 VWN n°5 SPWL 1992 Slater 0 Perdew Wang locale GGA
BLYP 1988 Becke88 0 Lee-Yang-Parr BP86 1988 Becke88 0 Perdew86 BPW91 1991 Becke88 0 Perdew-Wang 91 PBE 1996 Perdew-Burke-Ernzerhof 0 Perdew-Burke-Ernzerhof BPBE 1996 Becke88 0 Perdew-Burke-Ernzerhof G96LYP 1996 Gill96 0 Lee-Yang-Parr HCTH 1998 Hamprecht-Cohen-Tozer-Handy 0 Hamprecht-Cohen-Tozer-Handy mPWLYP 1998 modified Perdew-Wang91 0 Lee-Yang-Parr mPWPW91 1998 modified Perdew-Wang91 0 Perdew-Wang91 XLYP 2004 Becke88 + Perdew-Wang91 0 Lee-Yang-Parr MGGA BB95 1996 Becke88 0 Becke95 HGGA B3LYP 1994 Becke88 20 Lee-Yang-Parr HMGGA B1B95 1996 Becke88 25 Becke95 Une méthode ab initio ou semi-empirique ?
Les méthodes basées sur la théorie de la fonctionnelle de la densité sont considérées à l'heure comme une théorie ab initio par la plupart des scientifiques. En effet, les théorèmes de Hohenberg et Kohn ainsi que le développement amenant aux équations monoélectroniques de Kohn et Sham sont parfaitement rigoureux et sont obtenus sans avoir recours à d'éventuelles approximations. Cependant, la fonctionnelle d'échange-corrélation apparaissant dans les équations rend toutes résolutions exactes impossibles, sa forme analytique étant inconnue. Comme décrit plus haut, il est donc nécessaire d'approximer cette fonctionnelle soit en formulant une forme mathématique approchée de la fonctionnelle ou bien en fittant un certain nombre de données expérimentales. Cette approche est typique des méthodes semi-empiriques et la méthode DFT pourrait donc tout aussi bien être classée dans cette catégorie. L'énergie totale en DFT est exprimée en termes dépendant de la densité électronique plutôt qu'en termes de fonctions d'onde. Il est sans doute plus pertinent de considérer la méthode DFT comme une classe à part dans les méthodes numériques de chimie quantique.
Logiciels implémentant la DFT
Article détaillé : List of quantum chemistry and solid state physics software.- Abinit Site officiel
- ADF Site officiel
- AIMPRO Site officiel
- Atomistix Toolkit Site officiel
- CADPAC Site officiel
- CASTEP Site officiel
- CP2K Site officiel
- CPMD Site officiel
- CRYSTAL06 Site officiel
- DACAPO Site officiel
- DALTON Site officiel
- deMon2K Site officiel
- DFT++ Site officiel
- DMol3 Site officiel
- EXCITING Site officiel
- Fireball Site officiel
- FLEUR Site officiel
- GAMESS (UK) Site officiel
- GAMESS (US) Site officiel
- GAUSSIAN Site officiel
- GPAW Site officiel
- JAGUAR Site officiel
- MOLCAS Site officiel
- MOLPRO Site officiel
- MPQC Site officiel
- NWChem Site officiel
- OpenMX Site officiel
- ORCA Site officiel
- ParaGauss Site officiel
- PARATEC Site officiel
- PARSEC Site officiel
- PC GAMESS Site officiel
- PLATO Site officiel
- Parallel Quantum Solutions Site officiel
- PWscf Site officiel
- Q-Chem Site officiel
- SIESTA Site officiel
- Socorro Site officiel
- Spartan Site officiel
- SPR-KKR Site officiel
- TURBOMOLE Site officiel
- VASP Site officiel
- WIEN2k Site officiel
Notes et références
- (en) P.A.M. Dirac, « Quantum Mechanics of Many-Electron Systems », dans Proceedings of the Royal Society of London. Series A, Containing Papers of a Mathematical and Physical Character, vol. 123, no 792, 1929, p. 714-733 : The underlying physical laws necessary for the mathematical theory of a large part of physics and the whole of chemistry are thus completely known,and the difficulty is only that the exact application of these laws leads to equations much too complicated to be soluble.
- (en) E. Schrödinger, « An Undulatory Theory of the Mechanics of Atoms and Molecules », dans Phys. Rev., vol. 28, 1926, p. 1049-1070 [lien DOI]
- (fr)Il s'agit ici de l'équation de Schrödinger indépendante du temps.
- (de) M. Born et R. Oppenheimer, « Zur Quantentheorie der Molekeln », dans Annealen der Phyzik, vol. 389, 1927, p. 457-484 [lien DOI]
- (fr)La masse d'un nucléon est environ 1837 fois plus grande que la masse d'un électron
- (fr)Suite à l'approximation de Born-Oppenheimer, le hamiltonien total peut être séparé en un hamiltonien nucléaire et un hamiltonien électronique. Les propriétés des matériaux étant essentiellement caractérisées par le comportement des électrons, le hamiltonien décrit dans la suite de cet article sera toujours l'hamiltonien électronique.
- (fr) Par convention, la densité électronique est symbolisée par n(r) dans cet article. Il est cependant courant de la trouver symbolisée par ρ(r) dans de nombreux ouvrages et publications.
- (en) L.H. Thomas, « The calculation of atomic field », dans Proc. Cambridge Phil. Roy. Soc., vol. 23, 1927, p. 542-548
- (it) E. fermi, « Un metodo statistico per la determinazione di alcune priorieta dell'atome », dans Rend. Accad. Naz. Lincei, vol. 6, 1927, p. 602-607 [texte intégral]
- (en) Robert G. Parr et Weitao Yang, op. cit., 47
- (en) Norman H. March, op. cit, p. 24
- (en) P.A.M. Dirac, « Note on exchange phenomena in the Thomas-Fermi atom », dans Proc. Cambridge Phil. Roy. Soc., vol. 26, 1930, p. 376-385
- (de) C.F. von Weizsacker, « Zur Theorie der Kernmassen », dans Z. Phys., vol. 96, 1935, p. 431
- (en) R.P. Feynman, N. Metropolis et E. Teller, « Equations of state of elements based on the Thomas-Fermi theory », dans Phys. Rev., vol. 75, 1949, p. 1561-1573 [lien DOI]
- (en) E. Teller, « On the stability of molecules in the Thomas-Fermi theory », dans Rev. Mod. Phys., vol. 34, 1962, p. 627-630 [lien DOI]
- (en) P. Hohenberg et W. Kohn, « Inhomogenous Electron Gas », dans Phys. Rev., vol. 136, 1964, p. B864-B871 [lien DOI]
- (en) Richard L. Martin, op. cit. p. 135 : If you don't like the answer, change the question
- (en) W. Kohn, et L.J. Sham, « Self-Consistent Equations Including Exchange and Correlation Effects », dans Phys. Rev., vol. 140, 1965, p. A1133-A1138 [lien DOI]
- (en) Klaus Capelle, « A bird's-eye view of density-functional theory. », dans Braz. J. Phys., vol. 36, no 4A, 2006, p. 1318-1343 (ISSN 0103-9733)
- (fr) Contrairement à l'équation de Schrödinger, les valeurs propres des équations de Kohn-Sham ne représentent pas à proprement parler l'énergie.
- (fr) Bien qu'il soit commun de séparer ainsi l'échange et la corrélation, il existe néanmoins quelques doutes sur la légitimité de cette séparation.
- (en) Richard L. Martin, op. cit., p 120
- (en) J. C. Slater, « Simplification of the Hartree-Fock Method », dans Phys. Rev., vol. 81, 1951, p. 385-390 [lien DOI]
- (fr) Dans le cas de systèmes électroniques à couches fermées, la densité électronique des spin up et down sont égales et les approches LDA et LSDA deviennent identiques
- (en) D. Ceperley, « Ground state of the fermion one-component plasma: A Monte Carlo study in two and three dimensions », dans Phys. Rev. B, vol. 18, 1978, p. 3126-3138 [lien DOI]
- (en) D.M. Ceperley et B.J. Alder, « Ground State of the Electron Gas by a Stochastic Method », dans Phys. Rev. Lett., vol. 45, 1980, p. 566-569 [lien DOI]
- (en) R.L. Martin, op. cit. p. 479
- (en) J. P. Perdew and A. Zunger, « Self-interaction correction to density-functional approximations for many-electron systems », dans Phys. Rev. B, vol. 23, 1981, p. 5048-5079 [lien DOI]
- (en) S. H. Vosko, L. Wilk et M. Nusair, « Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis », dans Can. J. Phys, vol. 58, 1980, p. 1200-1211
- (en)Richard L. Martin, op. cit., p.479
- (en)Richard L. Martin, op. cit., p. 100
- (en)Richard L. Martin, op. cit., p. 479-480
- (en) Ref nécessaire : LSD is thus a first-principle approximation, in the sens that its parameters are not fitted empirically to calculated or experimentals results other than one in which its form is exact
- (en) A. D. Becke, « Density-functional exchange-energy approximation with correct asymptotic behavior », dans Phys. Rev. A, vol. 38, 1988, p. 3098 [lien DOI]
- (en) J.P. Perdew, K. Burke et Y. Wang, « Generalized gradient approximation for the exchange-correlation hole of a many-electron system », dans Phys. Rev . B, vol. 54, 1996, p. 16533-16539 [lien DOI]
- (en) C. Adamo et V. Barone, « Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models », dans J. Chem. Phys., vol. 108, 1998, p. 664 [lien DOI]
- (en) A.D. Becke, « Density-functional exchange-energy approximation with correct asymptotic behavior », dans Phys. Rev. A, vol. 38, 1988, p. 3098-3100 [lien DOI]
- (en) J.P. Perdew et Y. Wang, « Accurate and simple density functional for the electronic exchange energy: Generalized gradient approximation », dans Phys. Rev. B, vol. 33, 1986, p. 8800-8802 [lien DOI]
- (en) J.P. Perdew, K. Burke, et M Ernzerhof, « Generalized Gradient Approximation Made Simple », dans Phys. Rev. Lett., vol. 77, 1996, p. 3865-3868 [lien DOI]
- (en) Richard L. Martin op. cit. p 154-155
- (en) A.D. Becke, « Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing », dans J. Chem. Phys., vol. 104, 1996, p. 1040 [lien DOI]
- (fr) Ce choix est fondé sur le fait que l'échange électronique est décrit de manière exacte dans la théorie Hartree-Fock
- (en) S.F. Sousa, P.A. Fernandes et M.J. Ramos, « General Performance of Density Functionals », dans J. Phys. Chem. A, vol. 111, 2007, p. 10439-10452 [lien DOI]
- (en) (en) J.P. Perdew, K. Schmidt, Density Functional Theory and Its Application to Materials, V. Van Doren , C. Van Alsenoy , P. Geerlings, 1er août 2001, 220 p. (ISBN 9780735400160)
Annexes
Bibliographie
 : Ouvrage utilisé comme source pour la rédaction de cet article
: Ouvrage utilisé comme source pour la rédaction de cet article- (en) Robert G. Parr et Weitao Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, 1989, 350 p. (ISBN 0-19-509276-7)
- (en) Eberhard K.U. Gross et Reiner M. Dreizler, Density Functional Theory, Springer, 1995 (ISBN 0-306-44905-6)
- (en) Norman H. March, Electron Density Theory of Atoms and Molecules, Elsevier Science & Technology Books, 1997, 339 p. (ISBN 0-12-470525-1)
- (en) Carlos Fiolhais, Fernando Nogueira et Miguel Marques, A primer in Density Functional Theory, Springer, 2003, 256 p. (ISBN 3-540-03083-2)
- (en) Richard M. Martin, Electronic Structure Basic Theory and Practical Methods, Cambridge University Press, 2004, 648 p. (ISBN 0-521-78285-6)
Articles connexes
Liens externes
- [PDF] Electronic structure of Matter - Wave functions and density functionals : Nobel Lecture de Walter Kohn sur la DFT
 Portail de la chimie
Portail de la chimie Portail du monde quantique
Portail du monde quantique Portail de la physique
Portail de la physique
-




Wikimedia Foundation. 2010.