- Structure des proteines
-
Structure des protéines
La structure des protéines est la composition en acides aminés et la conformation en trois dimensions des protéines. Elle décrit la position relative des différents atomes qui composent une protéine donnée.
Les protéines sont des macromolécules de la cellule, dont elles constituent la « boîte à outils », lui permettant de digérer sa nourriture, produire son énergie, de fabriquer ses constituants, de se déplacer, etc. Elles se composent d'un enchaînement linéaire d'acides aminés liés par des liaisons peptidiques. Cet enchaînement possède une organisation tridimensionnelle (ou repliement) qui lui est propre. De la séquence au repliement, il existe 4 niveaux de structuration de la protéine.
Sommaire
Structure primaire
 Figure 1. Schéma général d'un acide aminé impliqué dans la structure primaire d'une protéine. R représente la chaîne latérale du résidu.
Figure 1. Schéma général d'un acide aminé impliqué dans la structure primaire d'une protéine. R représente la chaîne latérale du résidu.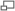
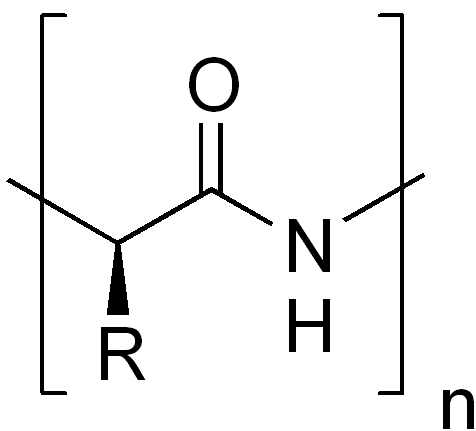
La structure primaire, ou séquence, d'une protéine correspond à la succession linéaire des acides aminés (ou résidus) la constituant. Les protéines sont donc des polymères d'acides aminés, reliés entre eux par des liaisons peptidiques.La structure primaire d'une protéine est le fruit de la traduction de l'ARNm en séquence protéique par le ribosome. C'est grâce au code génétique que l'information génétique (sous forme d'ARN) est traduite en acides aminés. Concrètement, la structure primaire est représentée par une succession de 20 lettres (code 1 lettre) différentes correspondant aux 20 acides aminés.
Nom code
3 lettrescode
1 lettreAbondance
relative
(%) E.C.M Chargé,
Polaire,
HydrophobeAlanine ALA A 13.0 71 H Arginine ARG R 5.3 157 C+ Asparagine ASN N 9.9 114 P Aspartate ASP D 9.9 114 C- Cystéine CYS C 1.8 103 P Glutamate GLU E 10.8 128 C- Glutamine GLN Q 10.8 128 P Glycine GLY G 7.8 57 - Histidine HIS H 0.7 137 P,C+ Isoleucine ILE I 4.4 113 H Leucine LEU L 7.8 113 H Lysine LYS K 7.0 129 C+ Méthionine MET M 3.8 131 H Phénylalanine PHE F 3.3 147 H Proline PRO P 4.6 97 H Sérine SER S 6.0 87 P Thréonine THR T 4.6 101 P Tryptophane TRP W 1.0 186 P Tyrosine TYR Y 2.2 163 P Valine VAL V 6.0 99 H Sélénocystéine SEC U rare - Pyrrolysine PYL J rare - Polarité, extrémités N- et C-terminales
La séquence primaire d'une protéine a un sens bien défini ou polarité. Le premier acide aminé de la séquence de la protéine est par convention celui qui possède une extrémité amine libre, on parle d'extrémité N-terminale ou de N-terminal. De manière symétrique le dernier acide aminé est celui qui possède une extrémité carboxylate libre, on parle de C-terminal.
Exemple d'une séquence d'acide aminé, l'α-lactalbumine humaine :
MRFFVPLFLVGILFPAILAKQFTKCELSQLLKDIDGYGGIALPELICTMFHTSGYDTQAI VENNESTEYGLFQISNKLWCKSSQVPQSRNICDISCDKFLDDDITDDIMCAKKILDIKGI DYWLAHKALCTEKLEQWLCEKL
Il existe des méthodes expérimentales de détermination de la structure primaire.
Structure secondaire
La structure secondaire décrit le repliement local de la chaîne principale[1] d'une protéine. L'existence de structures secondaires vient du fait que les repliements énergétiquement favorables de la chaîne peptidique sont limités et que seules certaines conformations sont possibles. Ainsi, une protéine peut être décrite par une séquence d'acides aminés mais aussi par un enchaînement d'éléments de structure secondaire.
De plus certaines conformations se trouvent nettement favorisées car stabilisée par des liaisons hydrogènes entre les groupements amide (-NH) et carbonyle (-CO) du squelette peptidique. Il existe trois principales catégories de structures secondaires selon l'échafaudage de liaisons hydrogènes, et donc selon le repliement des liaisons peptidiques : les hélices, les feuillets et les coudes.
Il existe des méthodes expérimentales pour déterminer la structure secondaire comme la résonance magnétique nucléaire, le dichroïsme circulaire ou certaines méthodes de spectroscopie infrarouge.
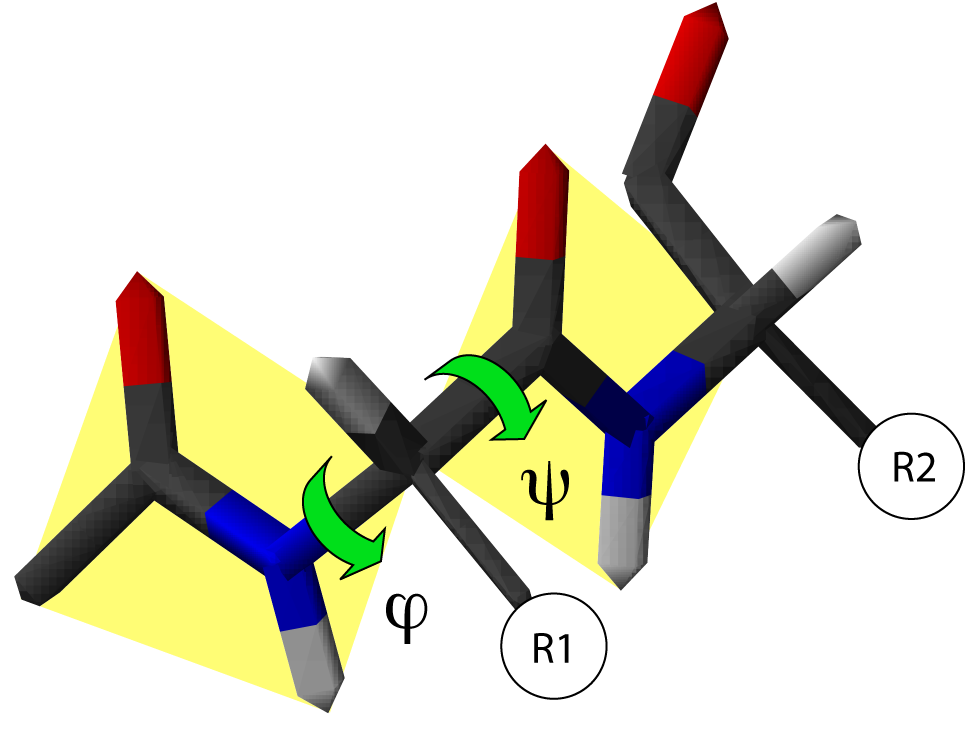
Angles dièdres et structure secondaire
 Angles dièdres dans une chaîne protéique. Il existe deux degrés de liberté de rotation, repérés par deux angles φ et ψ. Les liaisons peptidiques sont en jaune et R1 et R2 indiquent les chaînes latérales de deux acides aminés consécutifs
Angles dièdres dans une chaîne protéique. Il existe deux degrés de liberté de rotation, repérés par deux angles φ et ψ. Les liaisons peptidiques sont en jaune et R1 et R2 indiquent les chaînes latérales de deux acides aminés consécutifsLa chaîne principale contient trois liaisons covalentes par acide aminé. La liaison peptidique étant une liaison plane, il reste deux liaisons simples autour desquelles la rotation est possible. On peut donc déterminer la conformation du squelette d'un acide aminé à partir de deux angles dièdres, φ et ψ.
- L'angle dièdre φ est défini par les quatre atomes successifs du squelette : CO-NH-Cα-CO, le premier carbonyle étant celui du résidu précédent.
- L'angle dièdre ψ est défini par les quatre atomes successifs du squelette : NH-Cα-CO-NH, le second amide étant celui du résidu suivant.
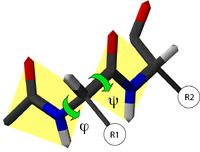
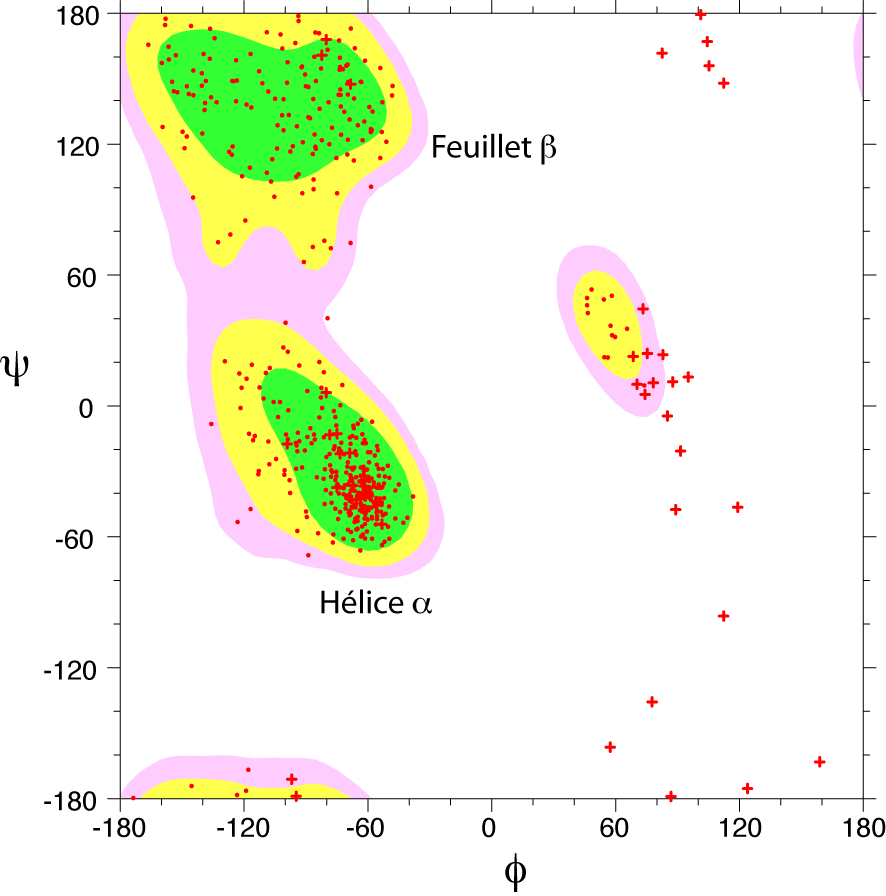
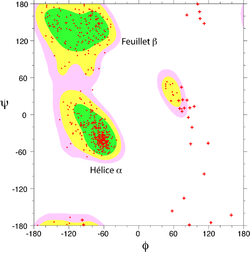 Diagramme de Ramachandran d'une protéine. Les zones énergétiquement favorables sont représentées par des contours colorés. Chaque acide aminé est représenté par un point rouge. Les croix, correspondent aux acides aminés glycine, qui ne comportent pas de chaînes latérales.
Diagramme de Ramachandran d'une protéine. Les zones énergétiquement favorables sont représentées par des contours colorés. Chaque acide aminé est représenté par un point rouge. Les croix, correspondent aux acides aminés glycine, qui ne comportent pas de chaînes latérales.Toutes les valeurs des angles φ et ψ ne sont pas possibles car certaines conduisent à des contacts trop proches entre atomes qui sont énergétiquement très défavorables. Une étude systématique des combinaisons admissibles d'angles φ et ψ a été réalisé par le biologiste et physicien indien Gopalasamudram Narayana Ramachandran en 1963[2]. Il a imaginé une représentation sous forme graphique de l'espace (φ ,ψ) qui porte aujourd'hui le nom de diagramme de Ramachandran. Ce diagramme montre trois principales zones énergétiquement favorables. Lorsqu'on analyse une structure de protéine, on observe que la majeure partie des acides aminés ont des combinaisons d'angles (φ ,ψ) qui s'inscrivent à l'intérieur de ces zones. Les deux principales régions correspondent aux structures secondaires régulières qui sont principalement observées dans les protéines : la région des hélices α et celle des feuillets β. La troisième région, plus petite, correspond à une conformation en hélice gauche (φ>0).
Il y a deux acides aminés particuliers qui font exception à cette règle du diagramme de Ramachandran : la Glycine et la Proline. La glycine ne possède pas de chaîne latérale (R=H) et, de ce fait, est beaucoup moins contrainte sur le plan de l'encombrement stérique. Elle peut donc adopter des valeurs (φ ,ψ) beaucoup plus diversifiées, en dehors des régions normalement privilégiées. À l'inverse, la proline est plus contrainte: elle contient un cycle pyrrole qui empêche la rotation correspondant à l'angle φ.
Hélice
Il y a conformation en hélice lorsque le squelette principal de la protéine adopte un repliement hélicoïdal périodique. Dans l'immense majorité des cas, cette hélice tourne dans le sens horaire. Elle est alors dite « droite ». Inversement, lorsqu'une hélice tourne dans le sens anti-horaire, elle est dite « gauche ».
Il existe aussi des enroulements superhélicoïdaux où 2 hélices, voire plus, s'enroulent l'une autour de l'autre pour former une superhélice. Ce type de conformation, ou faisceau d'hélices (coiled-coil) n'est pas une structure secondaire mais bien un type particulier de structure tertiaire, présent en particulier dans les protéines formant des fibres (e.g. fibrine, kératine, myosine).
Hélice α
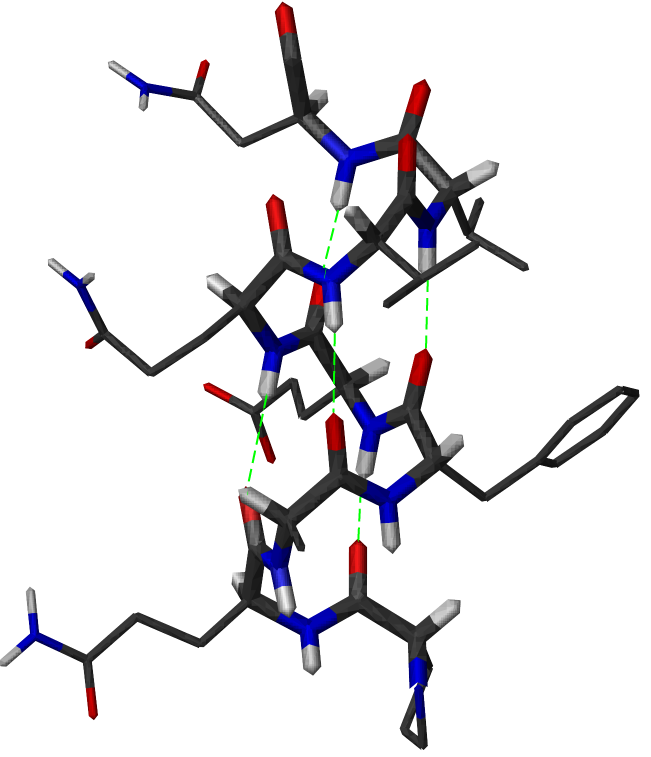
 Structure d'une hélice α. Les liaisons hydrogène sont figurées en vert. Le squelette principal est montré en tiges épaisses
Structure d'une hélice α. Les liaisons hydrogène sont figurées en vert. Le squelette principal est montré en tiges épaissesL'hélice α est une structure périodique très fréquente dans le repliement des protéines et des peptides. Elle se caractérise par la formation de liaisons hydrogènes entre le groupement carbonyle -CO d'un résidu i et le groupement amide -NH d'un résidu i+4. Un tour d'hélice α moyen contient 3,6 résidus et mesure 0.54 nm, soit une translation de 0.15 nm par résidu. Les angles dièdres φ et ψ de la chaîne peptidique sont en moyenne de -184° et -123° dans une hélice α.
Dans une hélice α, les chaînes latérales des acides aminés sont localisées en périphérie de l'hélice et pointent vers l'extérieur (voir figure). C'est une structure compacte, énergétiquement favorable.
La structure de l'hélice α a été prédite par Linus Pauling et Robert Corey en 1951[3], à partir de considérations théoriques, avant d'être observée effectivement pour la première fois en 1958 dans la myoglobine, la première protéine dont la structure tridimensionnelle a été résolue par cristallographie.
Hélice 310
L'hélice 310 se caractérise par la formation d'une liaison hydrogène entre le groupement -CO d'un résidu i et le groupement -NH d'un résidu i+3. Un pas d'hélice 310 moyen contient 3 résidus et mesure 0.60 nm, soit une translation de 0.2 nm par résidu. Les angles dièdres φ et ψ des liaisons peptidiques sont en moyenne de -49.0° et -26.0°. Le tour d'hélice 310 est donc plus étroit et plus contraint que celui de l'hélice α. Ce type de conformation est peu fréquent et sa longueur dépasse rarement 1 à 2 tours.
Hélice π
L'hélice π se caractérise par la formation d'une liaison hydrogène entre le groupement CO d'un résidu i et le groupement NH d'un résidu i+5. Un pas d'hélice π moyen contient 4 résidus et mesure 0.50 nm, soit une translation de 0.11 nm par résidu. Les angles dièdres φ et ψ des liaisons peptidiques sont en moyenne de -57.1° et -69.7°. Le tour d'hélice π est donc plus large que celui de l'hélice α. Ce type de conformation est très rare.
Hélice de type II
Les hélices de type II sont des hélices gauches formées par des poly-glycines ou des poly-prolines. Un pas moyen d'hélice de type II contient 3 résidus et mesure 0.93 nm, soit une translation de 0.31 nm par résidu. Les angles dièdres φ et ψ des liaisons peptidiques sont en moyenne de -79.0° et +145.0°.
Brin et feuillet β
Le brin β est une structure périodique étendue. Les liaisons hydrogènes qui le stabilisent se font entre résidus distants plutôt qu'entre résidus consécutifs, comme dans le cas de l'hélice α. En fait, un brin β seul n'est pas stable. Il a besoin de former des liaisons hydrogènes avec d'autres brins β pour se stabiliser. On parle alors de feuillets β. Un brin β est une structure de période 2, dont les chaînes latérales sont situées alternativement en dessus et en dessus du plan du feuillet. Grossièrement, le brin β peut être vu comme une hélice avec un pas de 2 acides aminés.
Les brins β composant un feuillet ont une polarité, celle de la chaîne peptidique qui va du N-terminal vers le C-terminal. Lors de l'agencement de deux brins adjacents dans un feuillet, deux topologies sont possibles : soit les deux brins ont la même orientation, soit ils ont des orientations opposées. Dans le premier cas, on parle de brins parallèles et dans le dernier de brins anti-parallèles.
Les feuillets β ne sont pas plans, ils présentent un plissement sur leur surface, avec des plis alternativement orientés vers le haut et vers le bas.
Feuillet β anti-parallèle
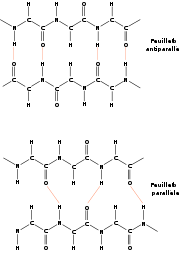 structures en feuillets β. En haut, feuillet anti-parallèle, en bas, feuillet parallèle
structures en feuillets β. En haut, feuillet anti-parallèle, en bas, feuillet parallèleLorsque les brins β s'organisent de manière tête-bêche, ils forment un feuillet β anti-parallèle. Les groupements -NH et -CO d'un résidu i d'un brin A forment des liaisons hydrogènes avec les groupements -CO et -NH d'un résidu j d'un brin B. Typiquement, 2 brins β consécutifs relié par un coude forment un feuillet β anti-parallèle. Un brin β moyen dans un feuillet anti-parallèle mesure 0.68 nm, soit une translation de 0.34 nm par résidu. Les angles dièdres φ et ψ des liaisons peptidiques sont en moyenne de -139.0° et +135.0°.
Feuillet β parallèle
Lorsque les brins β sont tous orientés dans le même sens, ils se forment en un feuillet β parallèle. Ainsi, 2 brins β consécutifs ne peuvent former un feuillet β parallèle. Les groupements -NH et -CO d'un résidu i d'un brin A forment des liaisons hydrogènes avec les groupements -CO d'un résidu et -NH d'un résidu j+2 appartenant à un brin B. Un brin β moyen dans un feuillet parallèle mesure 0.64 nm, soit une translation de 0.32 nm par résidu, Les angles dièdres φ et ψ des liaisons peptidiques sont en moyenne de -119.0° et +113.0°.
Un feuillet β est bien souvent composé de brins parallèles et anti-parallèles. Les feuillets β peuvent être plats mais ont plutôt tendance à former une structure légèrement gauche.
Coudes
Les coudes ne sont pas des structures périodiques. Il s'agit plutôt d'un repliement particulier du squelette carboné localisé à 3 ou 4 résidus consécutifs. Les coudes permettent bien souvent de relier 2 structures secondaires périodiques (hélices et/ou brins). Ils peuvent s'oxyder.
Coudes de type I, II et III
'Tableau 1 : Angles dièdres moyens des résidus centraux suivant les types de coudes. Résidu i+1 Résidu i+2 angle φ angle ψ angle φ angle ψ type I -60.0° -30.0° -90.0° 0.00° type II -60.0° +120.0° +80.0° 0.00° type III -60.0° -30.0° -60.0° -30.00° Tableau 2 :résidus favorisés suivant les types de coudes. Résidu i i+1 i+2 i+3 type I Asp, Arg,
Ser, CysPro tout sauf une Pro Gly type II Asp, Arg,
Ser, CysPro Gly, Arg Gly Dans les coudes de type I, II et III, il y a formation d'une liaison hydrogène entre le groupe -CO d'un résidu i et les groupement -NH d'un résidu i+3. Ces coudes courent donc sur 4 résidus. Ils sont regroupés sous l'appellation de coude β car ils font souvent le lien entre 2 brins β. Le tableau 1 récapitules les angles φ et ψ privilégiés des résidus au centre du coude.
Un coude de type III correspond à un tour d'hélice 310.
Certains acides aminés ont tendance à être favorisés à certaines positions des coudes selon leur encombrement stérique et/ou les angles dièdres qu'ils peuvent former (voir Tableau 2).
Il existe également des coudes de type I', II' et III' qui sont les images miroir des coudes décrit ci-dessus. Leurs angles dièdres sont les opposés de ceux décrits dans le Tableau 1.
Coude γ
Dans les coudes γ, il y a formation d'une liaison hydrogène entre le groupe -CO d'un résidu i et les groupement -NH d'un résidu i+2. Ces coudes courent donc sur 3 résidus. Les angles φ et ψ du résidu i+1 sont de +80.0° et -65.0°, respectivement. Il existe des coudes γ' avec des angles dièdres de -80.0° et +65.0°
Autres structures secondaires
Lorsque la conformation locale d'un segment protéique ne correspond à aucune de ces structures secondaires, on dit qu'il adopte une conformation en pelote statistique non périodique (random coil), par opposition aux hélices et aux feuillets qui sont des structures périodiques. Ce type de structure est le plus souvent associé aux boucles présentes entre 2 hélices ou feuillets. Pelote statistique ne signifie pas pour autant absence de structuration. Ainsi, certaines protéines ne possèdent aucun élément de structure secondaire régulière (hélice ou feuillet) mais ont une structure parfaitement stable. C'est souvent le cas des hormones et toxines polypeptidiques.
Structure tertiaire
La structure tertiaire d'une protéine correspond au repliement de la chaîne polypeptidique dans l'espace. On parle plus couramment de structure tridimensionnelle, ou structure 3D. La structure 3D d'une protéine est intimement liée à sa fonction: lorsque cette structure est cassée par l'emploi d'agent dénaturant, la protéine perd sa fonction: elle est dénaturée.
Dépendance de sa séquence
La structure tertiaire d'une protéine dépend de sa structure primaire. Ainsi, deux protéines homologues ayant une forte similiarité de séquence (> 80 % des acides aminés identiques) auront également des structures très proches. La prédiction de la structure tertiaire à partir de la structure primaire est à l'heure actuelle un champ très actif de la recherche en bio-informatique. Et de nombreuses méthodes utilisent justement l'homologie entre protéines pour réaliser leurs prédictions. Il est également connu de longue date que certains acides aminés favorisent la formation d'une structure secondaire plutôt qu'une autre[4]. Par exemple, la proline et la glycine ont une très faible propension a former des hélices α. En fait, de nombreuses méthodes bioinformatiques de prédiction de la structure tertiaire utilisent uniquement la séquence des protéines pour réaliser leurs prédictions.
Dépendance de son environnement
La structure tertiaire d'une protéine dépend aussi de son environnement. Les conditions locales qui existent à l'intérieur de chaque compartiment cellulaire, le solvant, la force ionique, la viscosité, la concentration, contribuent à moduler la conformation. Ainsi une protéine soluble dans l'eau aura besoin d'un environnement aqueux pour adopter sa structure tridimensionnelle. De même, une protéine membranaire aura besoin de l'environnement hydrophobe de la membrane pour adopter une conformation.
Effet hydrophobe[5]
La séquence d'une protéine comporte une certaine proportion d'acides aminés polaires (hydrophiles) et non polaires (hydrophobes). Leurs interactions avec les molécules d'eau conditionnent la manière dont la chaîne polypeptidique se replie. Les acides aminés non polaires auront tendance à éviter l'eau. Inversement les résidus polaires vont chercher a rester a proximité du solvant aqueux. Ainsi, dans le cas des protéines solubles, il se forme un cœur hydrophobe au centre de la structure tertiaire, tandis que les groupes polaires restent plutôt en surface.
Dans le cas des protéines transmembranaires le problème est inverse. L'environnement membranaire est globalement hydrophobe. Ainsi, les acides aminés hydrophiles vont se retrouver au cœur de la protéine tandis que les acides aminés hydrophobes vont se retrouver en surface. Des résidus hydrophiles peuvent se retrouver à la surface des protéines membranaires, en contact avec le milieu hydrophobe. Dans ce cas, il y a de forte chance que ces résidus soient impliqués dans des interactions avec d'autres résidus hydrophiles de la même ou d'une autre protéine.
Protéines intrinsèquement non-structurés
A l'heure actuelle, de plus en plus de chercheurs s'intéressent au cas des protéines intrinsèquement non structurées. Il s'agit de protéines généralement solubles n'ayant pas de structure 3D particulière sauf lorsqu'elle entrent en interaction avec d'autres facteurs : une autre protéine par exemple. En fait, ce type de protéines est souvent associée à plusieurs fonction biologiques, leur "souplesse" leur permettant de s'adapter à différentes interactions[6]. Les protéines intinsèquement non structurées représenteraient environ 10% des génomes[7]. Plus généralement, environ 40% des protéines eucaryotes possèderaient une région intrinsèquement non structurée[6]. Une base de donnée appelée Disprot fournit des informations supplémentaires sur ces protéines qui manquent de structure 3D fixe.Celle-ci répertorie toutes les structures intrinsèquement désordonnées connues jusqu'à présent et est régulièrement mise à jour.
Organisation
Les protéines s'organisent souvent en domaines structuraux distincts. Cela correspond aux parties de la protéine acquérant une conformation indépendamment du reste de la structure. Parfois ces domaines structuraux sont associés à une fonction individuelle donnée de la protéine : fixation d'un ligand, reconnaissance d'un autre partenaire, ancrage membranaire... Ainsi, une protéine constituée de plusieurs domaines structuraux peut associer plusieurs fonctions distinctes.
Il existe également dans les protéines des motifs structuraux. Ces motifs comptent quelques acides aminés et sont en général associés à des interactions bien précises. Par exemple, le motif « doigt de zinc » fixe l'ion Zn2+, qui est impliqué dans des interaction spécifiques avec l'ADN. Un motif peut être impliqué dans un domaine structural.
La représentation de la structure tertiaire se fait à l'aide d'un logiciel de visualisation comme Rasmol.
Lors de cette représentation, il est courant de faire apparaître certaines caractéristiques particulières des structures, comme les structures secondaires. Par exemple une hélice α sera représentée par un cylindre et les brins β qui composent les feuillets β par des rubans en forme de flèches, etc. La structure tertiaire est maintenue par différentes interactions:
- interactions covalentes (ponts disulfures entre cystéines)
- interactions électrostatiques (liaisons ioniques, liaisons hydrogènes)
- interactions de van der Waals
- interactions avec le solvant et l'environnement (ions, lipides...)
Détermination de la structure tertiaire
Différentes méthodes expérimentales permettent de découvrir la structure tertiaire des protéines :
- La cristallographie par rayon X
- La spectroscopie par résonance magnétique nucléaire
- La cryomicroscopie
Ces méthodes sont coûteuses et la détermination de la structure d'une protéine reste un processus lent. Afin de pallier ce défaut, des méthodes automatiques de prédiction de la structure tertiaire des protéines ont été développées. Il se dégage deux types de méthodes :
- Les méthodes basées sur les structures des protéines connues :
- Les méthodes par homologie
- Les méthodes par reconnaissance des repliements (Protein Threading)
- Les méthodes basées sur d'autres données (les propriétés physico-chimiques des atomes par exemple) :
- Les méthodes ab initio
- Les méthodes de novo
Structure quaternaire
La structure quaternaire des protéines regroupe l'association d'au moins deux chaînes polypeptidiques - identiques ou différentes - par des liaisons non-covalentes (liaison H, liaison ionique, interactions hydrophobes), mais rarement des ponts disulfures. L'effet hydrophobe est un facteur prépondérant dans l'assemblage des éléments structuraux, y compris dans l'association des sous-unités.
Chacune de ces chaînes est appelée monomère (ou sous-unité) et l'ensemble oligomère ou protéine multimérique.
L'hémoglobine est un exemple de structure quaternaire ; elle est constituée de 4 sous-unités : 2 sous-unités α (de 141 acides aminés) et 2 sous-unités β (de 146 acides aminés).Interactions responsables de la stabilité conformationnelle des protéines
Il est généralement admis que la structure d'une protéine "native" est thermodynamiquement la structure la plus stable. À l'exception des ponts disulfures qui n'existent que dans certaines protéines, principalement les protéines exocellulaires, les interactions qui stabilisent la conformation de ces molécules sont des interactions non covalentes. Toutes les interactions de ce type qui interviennent dans les petites molécules existent également dans les protéines. D'autre part, les interactions non covalentes ont lieu entre les divers groupes d'une protéine, mais aussi entre ces groupes et les molécules de solvant.
Ainsi l'énergie conformationnelle d'une molécule protéique est la somme de plusieurs contributions. Certaines de ces contribution résultent de facteurs intrinsèques à la protéine : ce sont les interactions de Van der Waals (non-bonded interactions) qui comportent un terme d'attraction et un terme de répulsion, les potentiels de torsions, les énergies de contraintes dans les angles ou les longueurs de liaison. D'autres proviennent d'interactions intramoléculaires influencées par le solvant, comme les liaisons hydrogène et les interactions électrostatiques. D'autres enfin sont principalement déterminées par le solvant, ce sont les interactions hydrophobes. Les liaisons hydrogène et les interactions hydrophobes présentent une dépendance de signe opposé par rapport à la température. Les liaisons hydrogène sont plus stables à basse température, à l'inverse des interactions hydrophobes; par suite la température correspondant au maximum de stabilité dépend de la proportion de ces interactions et par conséquent varie d'une protéine à l'autre. La structure native d'une protéine résulte d'un équilibre subtil entre différentes interactions stabilisantes et l'entropie conformationnelle qui tend à déstabiliser l'ensemble.
Recherche
Les projets suivants cherchent à mieux comprendre la structure des protéines :
- Docking@home (Université du Texas - El Paso)
- Folding@Home (Université Stanford)
- Proteins@home (École Polytechnique)
- Rosetta@home (Université de Washington)
- SPRINGS (Structural PRotein INformation Glossary Services) Université Pierre et Marie Curie (Institut de minéralogie et de Physique des Milieux Condensés), Université d'Arizona (Scientific Data Management Lab.) et Translational Genomics Research Institute (Pharmaceutical Genomics Division)
- RPBS (Ressource Parisienne en Bioinformatique Structurale)
Notes et références
- ↑ la chaîne principale d'une protéine correspond aux atomes impliqués dans la structure de base du polypeptide (-NH-CαH-CO-). Les chaînes latérales des acides aminés (souvent notées -R) n'appartiennent donc pas au squelette carboné.
- ↑ Ramachandran, G.N., Sasisekharan, V. & Ramakrishnan, C. (1963) J. Mol. Biol. 7, 95–99 (1963
- ↑ (en) L. Pauling, R.B. Corey, H.R. Branson. (1951) "The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain" Proc. Natl. Acad. Sci. USA, 37:205-211
- ↑ (en) p.ex. Argos P. and Palau J. (1982) Amino acid distribution in protein secondary structures, Int J Pept Protein Res, vol. 19(4):380-93.
- ↑ (en) p.ex. Richards F.M. and Richmond T. (1977) Solvents, interfaces and protein structure, Ciba Found Symp. vol. 60:23-45.
- ↑ a et b (en) Wright P.E. and Dyson H.J. (1999) Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm., J Mol Biol., vol. 293(2):321-31.
- ↑ (en) Tompa P. (2002) Intrinsically unstructured proteins, Trends Biochem Sci., vol. 27(10):527-533.
Bibliographie
- (fr) Lubert Stryer, Jeremy Mark Berg, John L. Tymoczko (trad. Serge Weinman), Biochimie, Flammarion, « Médecine-Sciences », Paris, 2003, 5e éd. (ISBN 2-257-17116-0).
- (fr) Carl-Ivar Brändén, John Tooze (trad. Bernard Lubochinsky, préf. Joël Janin), Introduction à la structure des protéines, De Boeck Université, Bruxelles, 1996 (ISBN 2-804-12109-7).
 Portail de la biochimie
Portail de la biochimie
Catégorie : Protéine

Wikimedia Foundation. 2010.