- PCR en temps reel
-
PCR en temps réel
Sommaire
Définition
La réaction en chaîne par polymérase en temps réel est une technologie ayant de nombreuses applications, basée sur une réaction enzymologique, la PCR et sur la mesure en continu de son produit.
Il existe différents appareils de PCR en temps réel.
A chaque cycle d’amplification, la quantité d’ADN total ou d’amplicon est mesurée grâce à un marqueur fluorescent. L’obtention de la cinétique complète de la réaction de polymérisation permet d’obtenir une quantification absolue de la quantité initiale d’ADN cible, ce qui était très difficile à obtenir sans biais en PCR en point final. Du point de vue enzymatique, il n’y a aucune différence théorique entre ces deux types de PCR. Vous êtes donc invité à consulter la page générale sur la Réaction en chaîne par polymérase . Grâce à l'extraordinaire puissance de la technique d'amplification de l'ADN par PCR, il est possible aujourd'hui d'établir un profil génétique à partir de quantités infimes d'ADN.Historique
- 1985 : Invention par Kary Mullis en 1985 de la technique de PCR (PCR).
- 1991 : première utilisation de sonde (sonde d’hydrolyse) en PCR publiée par Holland PM et collaborateurs.
- 1992 : première détection du produit de PCR en temps réel à l’aide d’un marquage au BET publiée par Higuchi R et collaborateurs.
Pourquoi quantifier en nombre de cycles?
La différence fondamentale de la PCR en temps réel avec la PCR en point final est que l’intégralité de la cinétique mesurable (au-dessus du bruit de fond) est quantifiée. Les données de fluorescence peuvent donc être exprimées en logarithme afin d’identifier facilement la phase exponentielle et mesurable, qui prend alors une apparence linéaire. Cette partie, alors appelée « segment quantifiable », permet de calculer la quantité d’ADN initial.
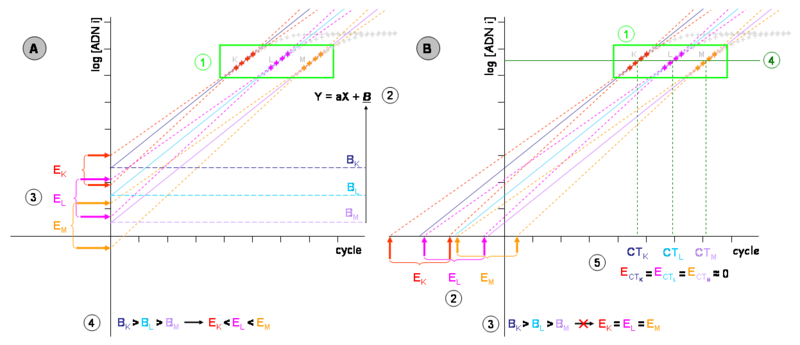
Graphique de gauche (A) :
- Les cinétiques de PCR théoriques de trois échantillons (K, L et M) de concentration d’ADN initial décroissantes (considérons d’un facteur 10 à chaque fois) sont représentées dans un repère semi logarithmique. Les mesures du bruit de fond ou trop influencées par lui (phase 1 et 2 d’une cinétique mesurable de PCR) ne sont pas représentées.
- (1) Zone des segments quantifiables de chaque échantillon (phase 3 d’une cinétique mesurable de PCR). Les autres phases sont représentées en gris et n’ont pas d’intérêt pour ce chapitre.
- (2) Chacun des segments quantifiables permet de définir une équation de type Y = aX + B. Cette équation de droite modélise la phase exponentielle de la PCR après transformation logarithmique, même la partie qui n’est pas mesurable à cause du bruit de fond. Les phases exponentielles des échantillons K, L et M sont représentées respectivement par les droites pleines bleu marine, turquoise et mauve. La pente « a » est dérivée de l’efficacité de PCR. Puisque le même amplicon est détecté pour chaque échantillon, c’est une constante et les droites sont parallèles. L’ordonnée à l’origine « B » correspond à la quantité d’ADN au cycle 0. La modélisation du segment quantifiable permet donc théoriquement de déterminer directement la quantité d’ADN initial.
- (3) En réalité, les mesures expérimentales ont toujours une erreur, aussi faible soit-elle, par définition imprédictibles et répondant à des phénomènes stochastiques. Si des échantillons de concentrations K, L et M étaient amplifiés plusieurs fois, on obtiendrait autant de cinétiques différentes, bien que très proches (leurs segments quantifiables sont respectivement représentés en rouge, rose et orange). Chacune de ces mesures permet d’établir une nouvelle équation de droite représentée en pointillés de la même couleur. Différents ordonnés à l’origine (B) sont alors mesurés. On considérera que les deux droites représentées pour chaque échantillon représentent l’erreur maximum de la technique. La différence entre leurs B (EK, EL et EM) représente donc l’incertitude de la mesure pour chaque échantillon. Cette incertitude est considérable. Dans l’exemple, elle est suffisante pour que l’on puisse mesurer L plus concentré qu K ou M que L mais en réalité, elle est souvent bien plus grande que cela (deux à quatre ordres de grandeur).
- (4) Remarquez que l’incertitude sur la mesure n’est pas identique pour chaque échantillon (EK est inférieure à EL, elle-même plus petite que EM). Une projection de l’équation de droite sur l’axe des ordonnées ou une de ses parallèles donne une incertitude concentration d’ADN initial dépendante.
Graphique de droite (B) :
- (1) Les équations déterminées à partir des segments quantifiables peuvent être extrapolées jusqu’à l’axe des abscisses, même si on obtient un point mathématique sans réalité biochimique.
- (2) Les droites modélisant l’erreur expérimentale (en pointillés et rouge, orange ou rose) permettent alors de définir de nouvelles incertitudes EK, EL et EM. Remarquez que ces incertitudes sont plus grandes que dans le graphique (A) mais de taille identique entre elles. L’erreur sur la mesure est donc devenue concentration d’ADN initial indépendante (3).
- (4) Il est possible de projeter les équations de droite sur une parallèle à l’axe des abscisses coupant les segments quantifiables par le milieu. Ce segment sera appelé « seuil de détection ».
- (5) Les valeurs en X (en nombre de cycles) de ces intersections sont généralement nommés CT (de l’anglais Cycle Threshold pour "cycle du seuil"), mais parfois aussi CP (de l’anglais Crossing Point pour "point de croisement"). Ce sont des valeurs mathématiques définies sur l’espace des réels positifs et non des entiers positifs (bien qu’une fraction de cycle n’ait aucune réalité expérimentale). Ces valeurs sont inversement proportionnelles à la quantité d’ADN initial, et l’incertitude sur la mesure est minimisée au maximum (en général inférieur à 5%).
La quantification en passant par une valeur mathématique en nombre de cycle (CT) permet donc d’obtenir des résultats fiables, mais n’est pas exploitable directement. Afin d’obtenir la quantité d’ADN initial, il va falloir réaliser de nouvelles transformations mathématiques qui nécessitent de connaître l’efficacité de PCR. Cette dernière est généralement déterminée grâce à une gamme d’étalonnage.
Gamme d'étalonnage
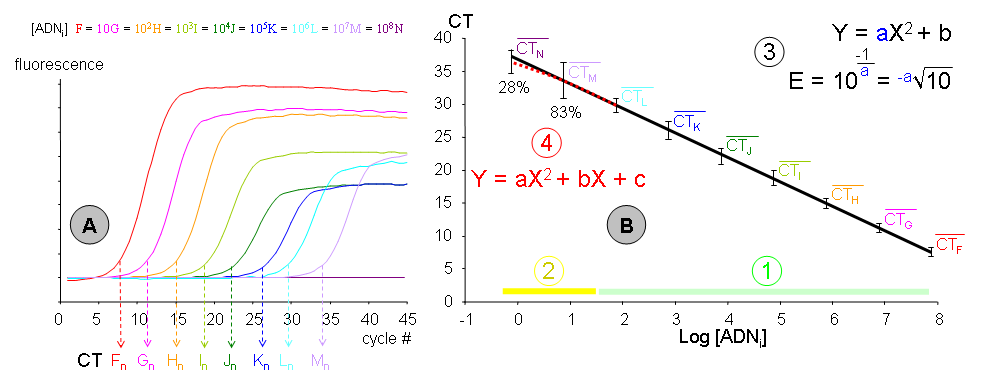
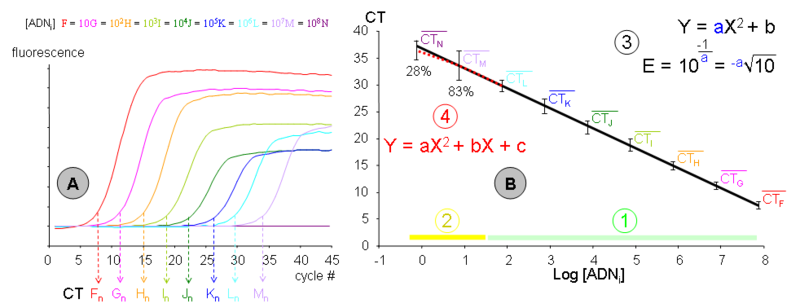 Graphique de gauche (A) :
Graphique de gauche (A) :- Neuf échantillons (F, G, H, I, J, K, L, M et N) de concentration en ADN initial décroissante (d’un ordre de grandeur à chaque fois) ont été amplifiés par PCR dans une même expérience. Chacune des cinétiques a permis de déterminer son CT propre, déterminé en nombre de cycle. Les concentrations en ADN sont connues en nombre de copies par tube (F ≈ 70 millions et N ≈ 0,7). Les données représentées correspondent à l’expérimentation « n », exemple parmi un grand nombre de dupliqués indépendants dans le temps, pour les lots des réactifs et pour les expérimentateurs. La fluorescence est exprimée en unités arbitraires et bruit de fond a été normalisé. Notez qu’aucune amplification n’a été obtenue pour l’échantillon Nn.
Graphique de droite (B) :
- Les moyennes des CT en fonction de la quantité d’ADN initiale de tous les dupliqués de gamme ont été reportées dans un repère semi-logarithmique. La droite d’étalonnage moyenne a été modélisée à l’aide d’une régression linéaire. La variabilité représentée n’est pas la SEM mais la dispersion (ou étendue) des mesures. Une gamme moyenne a été représentée afin d’illustrer les limites de la méthode, mais chaque gamme individuelle est néanmoins extrêmement robuste, avec le chiffre significatif du coefficient de détermination (r²) généralement à la quatrième décimale après la virgule (exemple : 0,9996) ! Il est donc utile de réaliser les droites d’étalonnage en PCR sur tableur, car les logiciels de la plupart des thermocycleurs sont beaucoup moins discriminants et ne peuvent modéliser qu’une gamme individuelle. Cette gamme moyenne fournit plusieurs informations :
- (1) La phase « détectable et quantitative de la PCR » : Tous les échantillons sont détectables et s’alignent avec les autres points de leur gamme individuelle. Cette phase comprend généralement des concentrations d’ADN initial allant de la centaine à la centaine de million de copies. En dessous, les phénomènes stochastiques deviennent très perceptibles mais peuvent être compensés par une multiplication des mesures. Au dessus, la phase de « bruit de fond » n’est plus assez importante pour pouvoir être correctement déterminée. Cela pourrait être compensé par un protocole de PCR ayant une efficacité très faible mais il est généralement plus simple de diluer l’échantillon.
- (2) La phase « parfois détectable mais non-quantitative de la PCR » : Elle comprend les concentrations d’ADN initial de l’ordre de la copie à la dizaine de copie. Le pourcentage d’échantillons détectés (pour cet exemple) a été indiqué pour les concentrations M et N, soit 83% pour une concentration moyenne de 7 copies et 28% lorsque trois tubes sur quatre contiennent une copie (concentration 0,7 ou -0,15 en log). La dispersion des mesures (et donc la marge d’erreur) devient beaucoup plus importante. Notez qu’un certain nombre de mesure M et N s’aligne convenablement avec leur gamme individuelle, mais ceci est du à la chance. Seule une multiplication des gammes permet donc de déterminer avec précision cette phase.
- (3) Une gamme étalon de PCR devenant une droite dans un repère semi-logarithmique, il est possible de la modéliser par une équation de type Y = aX + B où :
-
- Y est le CT mesuré par le thermocycleur.
- La pente (a) est une fonction de l’efficacité de PCR, cette dernière pouvant être calculée par l’équation:
-
![E = (indice\;du\;logarithme)^{\frac{-1}{pente}} = \sqrt[-pente]{indice\;du\;logarithme}](/pictures/frwiki/53/5c0af9783e3550961d2c1a82360ebff8.png) .
.-
-
- Notez que le schéma correspond au cas le plus fréquent où la concentration est exprimée en logarithme décimal (= d'indice 10). Cette pente est souvent considérée comme une constante pour l’amplification d’un amplicon particulier avec un protocole expérimental donné. La pente ou l’efficacité peuvent être employés pour quantifier les échantillons.
- X est la concentration d’ADN initial exprimée en log (de copies/tube, de ng/µl, d’unités arbitraires, etc.).
- B est un point mathématique qui n’a aucune réalité expérimentale (log 0 = 1/∞). Il peut néanmoins être utilisé pour calibrer les expériences de PCR (souvent nommée « run ») entre elles. Si les différentes gammes avaient été calibrées par leur intercept à l'origine (B), la dispersion serait apparue comme beaucoup plus faible, sauf pour les points M et N.
- Notez que le schéma correspond au cas le plus fréquent où la concentration est exprimée en logarithme décimal (= d'indice 10). Cette pente est souvent considérée comme une constante pour l’amplification d’un amplicon particulier avec un protocole expérimental donné. La pente ou l’efficacité peuvent être employés pour quantifier les échantillons.
- (4) La droite de régression ne passe pas au centre de la dispersion des différentes mesures pour les concentrations M et N, on constate un « amortissement » de la pente. Si l’on considère CT moyens L à N, ils seraient mieux modélisés par un polynôme du second degré. Certains logiciels associés aux thermocycleurs permettent de prendre en compte cet « amortissement ». Il convient cependant de noter que :
-
- Cet « amortissement » est extrêmement variable d’une gamme individuelle à une autre, et la correction modélisée ne correspond probablement pas à ce qui se passe dans l’échantillon quantifié. Les logiciels commerciaux modélisent cet « amortissement » sur une seule gamme, même si celle-ci comporte des biais évidents, et peuvent donc induire des erreurs très importantes que l’utilisateur novice ne décèlera pas forcément.
- L’imprécision sur la quantification à ces concentrations est tellement importante qu’une modélisation de « l’amortissement » est peu pertinente.
- Cet « amortissement » correspond, lorsque des gammes moyennes sont réalisées, à un abaissement de la pente, donc à une augmentation de l’efficacité de PCR, alors que l’accroissement des effets stochastiques devrait l’abaisser. Il faut noter alors que cet « amortissement » est généré principalement par la concentration la plus faible (N, soit 0,7 copies). Or aucune amplification ne pourra se faire s’il n’y a pas au moins une molécule d’ADN initial. Le biais qui en résulte dans la distribution gaussienne de l’erreur est susceptible de provoquer cet « amortissement moyen ».
-
-
Il faut avoir conscience que si la gamme d'étalonnage démontre l'aspect quantitatif du protocole expérimental, il est difficile d'éviter tous les biais potentiels, comme une différence de composition chimique entre le solvant des échantillons (milieu complexe d'ADN complémentaire, présence d'ARN, de protéines, etc.) et le diluant des points de la gamme (généralement de l'eau).
Transformation de CT (ou CP)
Le maxima de dérivé seconde
Applications
- mise au point d'amorces
- détection de mutations ponctuelles
- dosage d'OGM dans des produits pour la consommation humaine
- quantification
- détection spécifique et sensible de pathogènes d'intérêt vétérinaire - Quantification de la charge bactérienne, virale ou parasitaire
Bibliographie
- Elyse Poitras et Alain Houde (2002). La PCR en temps réel: principes et applications. Reviews in Biology and Biotechnology. 2(2):2-11.
- Bustin SA (2000). Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol. 25(2):169-93.
- Higuchi, R., Dollinger, G., Walsh, P.S., Griffith, R. (1992). Simultaneous amplification and detection of specific DNA-sequences. Bio-Technology 10 (4), 413–417.
- Holland, P.M., Abramson, R.D., Watson, R., Gelfand, D.H. (1991). Detection of specific polymerase chain reaction product by utilizing the 50 !30 exonuclease activity of Thermus aquaticus DNA polymerase. Proc. Natl. Acad. Sci. USA 88 (16), 7276–7280.
- Kubista M, Andrade JM, Bengtsson M, Forootan A, Jonak J, Lind K, Sindelka R, Sjoback R, Sjogreen B, Strombom L, Stahlberg A, Zoric N (2006). The real-time polymerase chain reaction. Mol Aspects Med.
Liens externes
- (en) http://www.gene-quantification.info Site contenant de nombreux liens et informations, associé à des références dans la tentative de minimisation du biais induit par l'utilisation de gène de ménage.
- (en) Guide pratique de PCR en temps réel (Université de Copenhague)
- (fr) Animation PCR en temps réel : principe de la PCR et de la PCR en temps réel et comparaison de ces techniques
courbe de fusion
 Portail de la biologie cellulaire et moléculaire
Portail de la biologie cellulaire et moléculaire Portail de la biochimie
Portail de la biochimie
Catégorie : Réaction en chaîne par polymérase

Wikimedia Foundation. 2010.