- Neurofibromatose de type ii
-
Neurofibromatose de type II
Cet article porte sur la neurofibromatose de type II qui n'a aucun rapport clinique et génétique avec la neurofibromatose de type I
Neurofibromatose de type II Autre nom Neurinome bilatéral de l'acoustique Référence MIM 101000 Transmission Dominante Chromosome 22q12.2 Gène NF2 Empreinte parentale Non Mutation Voir article Mutation de novo 50 % Nombre d'allèles pathologiques - Anticipation Non Porteur sain sans objet Incidence 1/25 000 Prévalence 1/100 000 Pénétrance 100 % Nombre de cas Inconnu Maladie génétiquement liée Aucune Diagnostic prénatal Possible Article principal Phacomatose Liste des maladies génétiques à gène identifié  Locus du gène responsable (rouge)
Locus du gène responsable (rouge)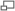
La neurofibromatose de type II est une maladie génétique se caractérisant par le développement de tumeurs bénignes : schwannomes vestibulaires bilatéraux.
Bien que ces tumeurs nerveuses soit bénignes, le nombre et la localisation de celles-ci entraînent une morbidité et une mortalité significative puisque l'âge moyen de décès est de 36 ans chez les individus atteints.
La neurofibromatose de type II est une phacomatose dont le mécanisme est un problème de différenciation du tissus ectodermique chez l'embryon.
Sommaire
Historique
La maladie a été décrite la première fois en 1822[1]. Son caractère héréditaire est noté en 1920[2]. Son gène a été identifié en 1993[3].
Génétique
Le gène NF2 est le seul gène connu responsable de neurofibromatose de type II. Plusieurs mécanismes sont responsables de son dysfonctionnement :
- Délétion majeure ou microscopique ;
- Chromosome 22 en anneau ;
- Translocation impliquant le chromosome 22.
Ce gène code pour une protéine, appelée merlin ou schwannomine, inhibant la croissance de certaines tumeurs et composée de 595 acides aminés.
La maladie est de transmission autosomique dominante, avec une pénétrance de près de 100% après l'âge de 60 ans[4].
Comme toute maladie génétique, elle peut être transmise aux descendants ou apparaître de novo par mutation. Si cette mutation a lieu lors de l'embryogenèse, seule une partie de l'individu est alors porteuse de celle-ci. On parle alors de mosaïque, donnant des formes partielles de la maladie. Cela serait le cas de près d'un tiers des malades sans antécédent familial, cette proportion pouvant doubler en cas de forme unilatérale de schwannome[5].
La mutation responsable peut être détectée chez l'individu asymptomatique (ne présentant aucun signe), en particulier chez les descendants d'un sujet atteint.
Épidémiologie
Elle concerne 1 naissance sur 25 000[4].
Description
atteinte neurologique
 Schwannome vestibulaire
Schwannome vestibulaireL'âge moyen d'apparition de la maladie est comprise entre 18 et 24 ans. Presque toutes les personnes atteintes développeront un schwannome vestibulaire devenant bilatérales avant l'âge de 30 ans. Les autres nerfs périphériques peuvent être atteints de schwannome ou de méningiome et rarement d'astrocytome ou épendymome. Une atteinte unique d'un nerf dans l'enfance responsable d'une paralysie faciale, d'un strabisme ou d'un pied et/ou d'une main ballante est un signe clinique de plus en plus fréquent vu dans l'enfance.
Le schwannome vestibulaire a pour conséquence une baisse de l'acuité auditive, qui est le premier symptôme, mais qui est retardé par rapport à l'apparition de la tumeur[6]. Elle peut être peu ressentie par le malade si l'atteinte est unilatérale, et devient très vite gênante si elle est bilatérale. L'évolutivité est cependant variable selon les individus et la taille de la tumeur n'est pas toujours correlée à l'importance de la gène[7]. Le schwannome peut atteindre également d'autres nerfs crâniens, ainsi que la moëlle épinière.
Un méningiome, tumeur bénigne des méninges, est présent dans environ la moitié des cas en intracrânien et au niveau de la moëlle épinière dans un cinquième des cas[4]. Ils sont souvent multiples. Leurs manifestations dépendent de leurs tailles et de leurs localisations. Ils sont détecté par un scanner crânien ou une IRM. La présence de cette tumeur est corrélée avec un plus mauvais pronostic[8].
L'épendymome,tumeur bénigne se développant au sein d'une des tuniques recouvrant la moëlle épinière, se voit également lors de cette maladie.
Il peut y avoir également atteinte nerveuse (neuropathie) sans objectivation d'une tumeur sur le nerf[9].
Atteinte oculaire
Une cataracte juvénile bien avant l'âge habituel de ce trouble de la vision, elle est présente chez près de deux tiers des sujets atteints de NF-2[10] et peut nécessiter une intervention chirurgicale.
Des hamartomes de la rétine sont décelés dans un cinquième des cas[11]. Des membranes, dites épirétiniennes, sont également fréquemment détectées[12].
Atteinte cutanée
Il existe fréquemment des tumeurs cutanées, souvent multiples, le plus souvent de type schwannome, parfois douloureuse ou sensible au toucher[13]. Des plaques de petites tailles, dépigmentées et glabres, ou au contraire hyperpigmentées et pileuses, se voient régulièrement dans cette maladie[4].
Critères diagnostics
Les critères diagnostics suivant, appelés « critères de Manchester » sont nécessaires pour établir précocement le diagnostic [14] :
- Schwannomes vestibulaires bilatéraux
- Un parent au premier degré atteint ET schwannome vestibulaire unilatéral OU deux anomalies suivantes : méningiome, schwannome, neurofibrome, gliome, cataracte
- Schwannome vestibulaire unilatéral ET deux anomalies suivantes : méningiome, schwannome, neurofibrome, gliome, cataracte
- Multiples meningiomes ET schwannome vestibulaire unilatéral OU deux anomalies suivantes : schwannome, neurofibrome, gliome, cataracte
Gravité
 Méningiome
MéningiomeCe qui caractérise essentiellement la NF-2, c'est le développement de neurinomes bilatéraux (schwannomes) de la VIIIe paire de nerfs crâniens (retrouvés dans plus de 90 % des cas).
Révélés vers l'âge de 20-30 ans, ils constituent le problème majeur de la NF-2 par le retentissement sur l'ouïe, l'équilibre et aussi si leur développement comprime d'autres structures cérébrales.
La durée moyenne de vie est diminuée à environ 60 ans[4].
Traitement
Il n'existe actuellement aucun traitement spécifique.
Le traitement du schwannome vestibulaire est l'ablation complète de la ou des tumeurs. Parfois l'ablation est partielle, et associée à de la radiothérapie et de la radio chirurgie, mais le risque de récidive est alors plus important.
L'intervention est souvent complexe car la tumeur est assez adhérente aux tissus voisins et englobe parfois de nombreux faisceaux nerveux[15]. Les résultats sont parfois incomplets quant à la récupération de l'acuité auditive[16].
La mise en place d'un implant cochléaire peut être proposée si la cochlée est intacte, avec son nerf[17].
Les méningiomes sont, par contre, bien séparés des structures voisines et leur ablation chirurgicale ne pose pas de gros problèmes, sauf s'ils sont très proches de certaines structures cérébrales délicates d'accès ou fragiles (base du crâne en particulier, localisation qui correpond à l'émergence des nerfs crâniens[18]).
Sources
- (en) D Gareth Evans, Neurofibromatosis 2 In : GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997-2006. [1].
- ↑ Wishart JH, Case of tumours in the skull, dura mater and brain, Edinburgh Med Surg J, 1822;18:393-397
- ↑ Feiling A, Ward E, A familial form of acoustic tumour, BMJ, 1920;10:496-497
- ↑ Rouleau GA, Merel P, Lutchman M et als. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro-fibromatosis type 2, Nature, 1993;363: 515-521
- ↑ a , b , c , d et e Asthagiri AR, Parry DM, Butman JA et Als. Neurofibromatosis type 2, Lancet, 2009;373:1974-1986
- ↑ Evans DGR, Ramsden RT, Shenton A et als. Mosaicism in neurofibromatosis type 2: an update of risk based on uni/bilaterality of vestibular schwannoma at presentation and sensitive mutation analysis including multiple ligation-dependent probe amplification, J Med Genet, 2007;44:424-428
- ↑ Masuda A, Fisher LM, Oppenheimer ML, Iqbal Z, Slattery WH, Hearing changes after diagnosis in neurofibromatosis type 2, Otol Neurotol, 2004;25:150-154
- ↑ Baser ME, Mautner VF, Parry DM, Evans DGR, Methodological issues in longitudinal studies: vestibular schwannoma growth rates in neurofibromatosis 2, J Med Genet, 2005;42:903-906
- ↑ Baser ME, Friedman JM, Aeschliman D et als. Predictors of the risk of mortality in neurofibromatosis 2, Am J Hum Genet, 2002;71:715-723
- ↑ Sperfeld AD, Hein C, Schroder JM, Ludolph AC, Hanemann CO, Occurrence and characterization of peripheral nerve involvement in neurofibromatosis type 2, Brain, 2002;125:996-1004
- ↑ Bosch MM, Boltshauser E, Harpes P, Landau K, Ophthalmologic findings and long-term course in patients with neurofibromatosis type 2, Am J Ophthalmol, 2006;141:1068-1077
- ↑ Ragge NK, Baser ME, Klein J et als. Ocular abnormalities in neurofibromatosis 2, Am J Ophthalmol, 1995;120:634-641
- ↑ Kaye LD, Rothner AD, Beauchamp GR, Meyers SM, Estes ML, Ocular findings associated with neurofibromatosis type II, Ophthalmology, 1992;99:1424-1429
- ↑ Mautner VF, Lindenau M, Baser ME, Kluwe L, Gottschalk J, Skin abnormalities in neurofibromatosis 2, Arch Dermatol, 1997;133:1539-1543
- ↑ Evans DGR, Baser ME, O'Reilly B et als. Management of the patient and family with neurofibromatosis 2: a consensus conference statement, Br J Neurosurg, 2005;19:5-12
- ↑ Jaaskelainen J, Paetau A, Pyykko I, Blomstedt G, Palva T, Troupp H, Interface between the facial nerve and large acoustic neurinomas. Immunohistochemical study of the cleavage plane in NF2 and non-NF2 cases, J Neurosurg, 1994;80:541-547
- ↑ Brackmann DE, Fayad JN, Slattery Iii WH et als. Early proactive management of vestibular schwannomas in neurofibromatosis type 2, Neurosurgery, 2001;49:274-283
- ↑ Hoffman RA, Kohan D, Cohen NL, Cochlear implants in the management of bilateral acoustic neuromas, Am J Otol, 1992;13:525-528
- ↑ Larson JJ, Van Loveren HR, Balko MG, Tew JM, Evidence of meningioma infiltration into cranial nerves: clinical implications for cavernous sinus meningiomas, J Neurosurg, 1995;83:596-599
 Portail de la médecine
Portail de la médecine
Catégorie : Maladie génétique


Wikimedia Foundation. 2010.