- Méthodes de chimie numérique dans les systèmes solides
-
Chimie numérique
La chimie numérique (désignée parfois aussi par l'anglicisme chimie computationnelle) est une branche de la chimie et/ou de la physico-chimie qui utilise les lois de la chimie théorique exploitées dans des programmes informatiques spécifiques afin de calculer structures et propriétés d'objets chimiques (molécules, solides, clusters, surfaces ou autres), en appliquant autant que possible ces programmes à des problèmes chimiques réels. La frontière entre la simulation effectuée et système réel est bien sûr définie par le niveau de précision requis et/ou la complexité des systèmes étudiés et les théories employées lors de la modélisation. Les propriétés recherchées peuvent être la structure (géométrie, relations entre constituants), l'énergie totale, l'énergie d'interaction, les charges, dipôles et moments multipolaires, fréquences vibrationnelles, réactivité ou autres quantités spectroscopiques, sections efficaces pour les collisions, etc. La sous-discipline la plus représentée est celle qui s'applique au traitement des configurations électroniques des systèmes.
L'expression chimie numérique est parfois également utilisée pour désigner tous les champs scientifiques qui recouvrent à la fois la chimie et l'informatique.Introduction
Le terme de chimie théorique peut être défini comme la description mathématique de la chimie, tandis que la chimie numérique est habituellement utilisée lorsqu'une méthode mathématique est suffisamment bien développée pour être automatisée puis implémentée dans un code de calcul. Il convient de remarquer que les mots exact et parfait n'apparaissent pas ici, peu de quantités chimiques pouvant être calculées de manière exacte. Cependant, chaque propriété chimique peut être décrite par un schéma numérique de manière qualitative ou de manière quantitative approximative.
Les molécules (et plus précisément les atomes qui les constituent) sont constituées de noyaux et d'électrons, ce qui fait que les méthodes de la chimie quantique peuvent s'appliquer. Les chimistes numériciens essaient parfois de résoudre l'équation de Schrödinger non relativiste, avec ajout de corrections relativistes, bien que des progrès aient été réalisés dans la résolution de l'équation de Schrödinger totalement relativiste. Il est, en principe, possible de résoudre exactement l'équation de Schrödinger, soit dans sa forme dépendante soit dans sa forme indépendante du temps, mais cela n'est possible en pratique que pour des systèmes très petits. Cependant, de nombreuses méthodes d'approximations permettent d'obtenir le meilleur compromis entre précision et « coût numérique ». La chimie numérique actuelle peut permettre de manière routinière et très précise les propriétés de systèmes chimiques contenant jusqu'à une quarantaine d'électrons au plus[1]. Le traitement de systèmes plus importants (quelques dizaines d'électrons) sont traitables de manière numérique par des méthodes d'approximation comme la théorie de la fonctionnelle de la densité (DFT). Il existe une controverse dans la discipline sur la pertinence et la précision des dernières méthodes pour décrire des réactions chimiques complexes, comme par exemple en biochimie. Les systèmes complexes peuvent être étudiés au moyen de méthodes semi-empiriques - se distinguant des précédentes dites ab initio - qui traitent les interactions en les « lissant » sur des résultats théoriques ou ... ab initio. Le niveau d'approximation encore supérieur, qui permet de traiter les systèmes les plus importants (comme des chaînes d'ADN ou autres membranes cellulaires par des méthodes relevant de la mécanique classique, appelée alors mécanique moléculaire (classique)[2].En chimie théorique, les chimistes, les physiciens et les mathématiciens développent des algorithmes et des codes afin de prédire des propriétés atomiques, moléculaires ou autres, et éventuellement des chemins de réactions chimiques. Les chimistes numériciens, a contrario, peuvent appliquer simplement les codes et méthodologies existants pour des problématiques chimiques spécifiques. Il existe deux aspects distincts de la chimie numérique :
- les études menées pour trouver un point de départ pour une synthèse de laboratoire, ou pour expliciter des résultats expérimentaux, comme la position et la source des pics spectroscopiques.
- les études menées pour prédire la possibilité d'existence pour des systèmes inconnus ou d'explorer des mécanismes réactionnels qui ne peuvent être étudiés par des moyens expérimentaux.
La chimie numérique se place donc à la fois en amont (moyen de prédiction et de prospection) et en aval (moyen d'explication) de l'expérience [3]. Plusieurs secteurs majeurs de la chimie numérique peuvent être distingués :
- la prédiction de structures moléculaires, cristallines, ou autres états stables ou métastables de systèmes physico-chimiques par la détermination des forces appliquées afin de trouver les points stationnaires de l'hypersurface d'énergie lorsque la position des noyaux (ou ions) varie par exemple, ou lorsque les états d'excitation électroniques sont relaxés.
- l'accumulation et la recherche de données sur des systèmes chimiques (base de données chimiques).
- l'identification les corrélations entre structure chimique et propriétés démontrées par le système étudié (voir QSPR / QSAR)
- les approches numériques d'aide à la synthèse de différents composés.
- les approches numériques à la conception de systèmes chimiques interagissant de manière spécifique avec d'autres systèmes (comme par exemple la conception de médicaments).
Historique bref
Construits sur les découvertes fondatrices et les théories énoncées durant l'histoire de la chimie quantique, les premiers calculs théoriques en chimie ont été produits par Walter Heitler et Fritz London en 1927. Dans cette perspective, quelques livres furent grandement influents dans le développement initial de la chimie numérique quantique :
- Introduction to Quantum Mechanics – with Applications to Chemistry de Pauling et Wilson (1935).
- Quantum Chemistry de Eyring, Walter et Kimball (1944).
- Elementary Wave Mechanics – with Applications to Quantum Chemistry de Heitler (1945).
- Valence de Coulson (1952).
Chacun d'entre eux servit de référence primaire pour les chimistes dans les décennies qui suivirent.
Avec le développement de l'informatique dans les années 1940, le cacul par ordinateur des solutions de l'équation d'onde pour des complexes atomiques est devenu un objectif réalisable. Au début des années 1950, les premiers calculs d'orbitales semi-empiriques furent menés. Les chimistes théoriciens devinrent des utilisateurs intensifs des premiers ordinateurs. Un compte rendu très détaillé d'une telle utilisation est donné par Smith et Sutcliffe [4]. Les premiers calculs de type Hartree-Fock ab initio sur des molécules diatomiques furent effectués en 1956 au MIT avec une base d'orbitales de Slater. Pour des molécules diatomiques, une étude systématique utilisant une base minimale et un premier calcul utilisant une base plus importante furent publiés respectivement par Ransil et Nesbet en 1960[5].
Les premiers calculs polyatomiques utilisant des orbitales gaussiennes ont été menés à la fin des années 1950. Les premiers calculs d'interaction de configuration ont été effectués à Cambridge sur le calculateur EDSAC II dans les années 1950 au moyen d'orbitales gaussiennes également, par Boys et collaborateurs [6]. En 1971, lorsqu'une première bibliographie de calculs ab initio fut publiée[7], les plus grosses molécules qui y étaient citées étaient le naphtalène et l'azulène [8],[9]. Un résumé des développements antérieurs a été publié par Schaefer[10].
En 1964, la méthode de Hückel, méthode de LCAO simple pour la détermination des énergies électroniques des orbitales moléculaires des électrons π dans les hydrocarbures conjugués, allant d'objets simples comme le butadiène ou le benzène à l'ovalène avec dix cycles à six atomes accolés, fut utilisée sur les ordinateurs de Berkeley et Oxford[11]. Ces méthodes empiriques furent peu à peu remplacées dans la même décennie par des méthodes semi-empiriques comme la méthode CNDO[12]. Au début des années 1970, des programmes informatiques (relativement) efficaces comme ATMOL, POLYATOM, IBMOL, et GAUSSIAN, etc. ont commencé à être utilisés pour accélérer les calculs sur les orbitales moléculaires. Dans le même temps, les méthodes de mécanique moléculaire, comme MM2, étaient développées, en premier lieu par Norman Allinger [13].Une des premières mentions du terme de « chimie numérique » peut être trouvée dans le livre Computers and Their Role in the Physical Sciences (1970) de Sidney Fernbach et Abraham Haskell Taub, où ils indiquent « il semble, à présent, que la chimie numérique peut finalement être de plus en plus tangible »[14]. À partir de ces premiers développements, la chimie numérique scientifique émergea comme discipline distincte en 1979 environ[15]. Durant les années 1980, les bases de la chimie numérique furent établies. Des publications spécifiquement dédiées virent le jour par la suite, comme le Journal of Computational Chemistry, par exemple, qui fut pour la première fois édité en 1980.
Études des structures moléculaires
Une formule moléculaire donnée peut représenter en réalité de nombreux isomères. Chacun d'entre eux représente un minimum local de la surface d'énergie (appelée surface d'énergie potentielle) créée à partir de l'énergie totale (énergie électronique, énergie de répulsion entre noyaux, etc.) en tant que fonction des coordonnées de l'ensemble des noyaux. Un minimum local (d'énergie) est un point stationnaire à partir duquel tout déplacement conduit à un accroissement de l'énergie totale du système. Le minimum local le plus bas est appelé minimum global et correspond à l'isomère le plus stable pour la formulation initiale. S'il existe une modification de coordonnées particulière qui conduit à une décroissance de l'énergie dans les deux directions, le point stationnaire correspond à une structure de transition et la coordonnée est la coordonnée de réaction. Le procédé de recherche des points stationnaires est appelé optimisation de géométrie[16].
La détermination d'une structure moléculaire par optimisation de géométrie devient routinière lorsque des méthodes de calcul des dérivées premières de l'énergie selon toutes les coordonnées atomiques efficaces sont disponibles. L'évaluation des dérivées secondes liées permet de prédire les fréquences vibrationnelles si un mouvement harmonique est supposé. Cela permet, de manière simplifiée, de caractériser les points stationnaires. Les fréquences sont liées aux valeurs propres de la matrice des dérivées secondes (matrice hessienne). Si les valeurs propres sont toutes positives, alors les fréquences sont toutes réelles et le point stationnaire est un minimum local. Si l'une d'entre elles est négative, c'est-à-dire correspond à une fréquence imaginaire, le point stationnaire correspond à une structure de transition. Si plus d'une d'entre elles est négative, le point stationnaire est plus complexe, et habituellement de peu d'intérêt. Lorsque ce type de point est découvert, il est nécessaire de déplacer la recherche vers d'autres points si l'on recherche les minima locaux et les structures de transition.
L'énergie totale est déterminée par les solutions approchées de l'équation de Schrödinger dépendante du temps, habituellement avec les termes non relativistes inclus, et en utilisant l'approximation de Born-Oppenheimer qui, en se basant sur la plus grande vélocité des électrons comparée à celle des noyaux, permet la séparation (découplage) des mouvements électroniques et nucléaires, et simplifie de fait l'équation de Schrödinger. Ceci permet d'évaluer l'énergie totale comme une somme de l'énergie électronique pour des positions nucléaires fixes et de l'énergie de répulsion des noyaux. Une exception notable est développée dans certaines approches connues sous le nom générique de chimie quantique directe, qui traitent les électrons et les noyaux sur un pied d'égalité. Les méthodes basées sur la fonctionnelle de la densité et les méthodes semi-empiriques peuvent être considérées comme des variantes sur ce thème majeur. Pour des systèmes très étendus, l'énergie totale est déterminée par mécanique moléculaire. Les différentes techniques pour prédire l'énergie totale des structures moléculaires sont :
Les méthodes ab initio
Article détaillé : Méthode ab initio de chimie quantique.Les codes utilisés en chimie numérique moléculaire sont basés sur de nombreuses et différentes méthodes de chimie quantique qui permettent la résolution de l'équation de Schrödinger associée au hamiltonien moléculaire. Les méthodes qui n'incluent aucun paramètre empirique ou semi-empirique dans leurs équations, c'est-à-dire qui dérivent directement des principes théoriques, sans inclusion de données expérimentales, sont appelées méthodes ab initio. Cela n'implique pas que la solution obtenue est la solution exacte; elles consistent toutes en des approximations à divers degrés des calculs de mécanique quantique[17]. Cela signifie qu'une approximation particulière est définie de manière rigoureuse sur les premiers principes (théorie quantique) puis résolue avec une marge d'erreur qui est connue de manière qualitative à l'avance. Si des méthodes par itérations numériques sont utilisées, le but est d'itérer jusqu'à atteindre la précision machine[18].
 Diagramme illustrant diverses méthodes ab initio en termes d'énergies.
Diagramme illustrant diverses méthodes ab initio en termes d'énergies.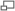
La méthode ab initio la plus simple de calcul de structure électronique est le schéma Hartree-Fock (HF), dans laquelle la répulsion coulombienne électron-électron n'est pas spécifiquement prise en compte. Seul son effet moyen est inclus dans le calcul. Lorsque la taille de la base est augmentée, l'énergie et la fonction d'onde tendent vers une limite appelée limite Hartree-Fock. De nombreux types de calculs, connus sous le nom de méthodes post-Hartree-Fock, commencent par un calcul Hartree-Fock et sont ensuite corrigés pour la répulsion électron-élection, aussi connue comme corrélation électronique. Lorsque ces méthodes sont poussées vers leurs limites, elles approchent[19] de la solution exacte de l'équation de Schrödinger non-relativiste. Si l'on veut obtenir un accord exact avec l'expérience, il est nécessaire d'inclure les termes relativiste et de spin-orbit, les deux n'étant importants uniquement pour les atomes lourds. Dans toutes ces approches, en plus du choix de la méthode, il est nécessaire de choisir une base adéquate. Cette base (au sens mathématique du terme) est un ensemble de fonctions, habituellement centrées sur les différents atomes de la molécule[20], qui sont utilisées pour étendre les orbitales moléculaires par le postulat[21] de combinaison linéaire d'orbitales atomiques (LCAO). Les méthodes ab initio nécessitent donc d'établir un niveau d'application de la théorie (méthode) et une base.
La fonction d'onde Hartree-Fock est une configuration ou un déterminant simple. Dans certains cas, particulièrement dans les processus de ruptures de liaisons chimiques, elle est relativement inadéquate et de nombreuses configurations sont alors nécessaires. Dans ce cas, les coefficients des configurations et les coefficients des fonctions de base sont optimisés de concert.
L'énergie moléculaire totale peut être évaluée comme une fonction de la géométrie moléculaire, c'est-à-dire, en d'autres termes, la surface d'énergie potentielle.
Exemple : Si2H2 ressemble-t-il à C2H2 ?
Des séries d'études ab initio sur Si2H2 (disyline) ont clairement montré la pertinence de la chimie numérique ab initio. Elles s'étendirent sur presque 20 ans, et les conclusions principales furent publiées en 1995. Les méthodes utilisées étaient essentiellement post-Hartree-Fock, en particulier les méthodes d'interactions de configuration (CI) et de clusters couplés (CC). La question initiale était de savoir si Si2H2 possédait la même structure que l'éthyne (acétylène) C2H2. Peu à peu (cette étude ayant commencé avant que l'optimisation de géométrie soit largement répandue), il devint clair que Si2H2 linéaire était en fait une structure de transition entre deux structures trans-déformées équivalentes, et qu'il était en fait moins stable (plus haut en énergie). L'état fondamental fut prédit comme un cycle à quatre déformé en une structure « papillon » avec des atomes d'hydrogènes reliés entre les deux atomes de silicium. L'intérêt dévia ensuite vers le fait de savoir si des structures équivalentes au vinylidène c'est-à-dire Si=SiH2, existent. Cette structure est calculée comme étant un minimum local, soit un isomère de Si2H2, plus élevé en énergie que l'état fondamental mais plus bas en énergie que l'isomère trans-déformé. Puis, de manière surprenante, un nouvel isomère fut prédit par Brenda Colegrove, de l'équipe d'Henry F. Schaefer[22]. Cette découverte fut si étonnante qu'elle nécessita des calculs extensifs pour être confirmée. Il est en effet nécessaire de recourir à des méthodes post-Hartree-Fock afin d'obtenir le minimum local correspondant à cette structure. En effet, ce minimum n'existe tout bonnement pas sur l'hypersurface d'énergie Hartree-Fock. Ce nouvel isomère est une structure plane avec un atome d'hydrogène ponté et un atome d'hydrogène terminal, en position cis par rapport à l'atome ponté. Son énergie est au-dessus de celle de l'état fondamental mais inférieure à celle des autres isomères[23] Des résultats similaires ont été obtenus pour Ge2H2 [24], le germanium appartenant à la même colonne que le silicium dans la classification périodique. De façon peut être plus intéressante, des résultats similaires ont aussi été obtenus pour Al2H2[25] (puis pour Ga2H2)[26] qui a globalement deux électrons de valence en moins par rapport aux molécules constituées à partir d'éléments du groupe 14[27]. La seule différence est que l'état fondamental du cycle à quatre atomes est plan et non plus déformé. Les isomères cis-mono-pontés et « vinylidène » sont aussi présents. Le travail expérimental sur ces molécules n'est pas aisé, mais la spectroscopie par isolation de matrice des produits de réaction d'atomes d'hydrogène sur des surfaces de silicium et d'aluminium a permis de retrouver les structures cycliques des états fondamentaux ainsi que les structures cis-mono-pontées pour Si2H2 et Al2H2. Les prédictions théoriques sur les fréquences vibrationnelles sont cruciales pour comprendre les observations expérimentales des spectres d'un mélange de composés. Ceci peut apparaître comme un pan obscur de la chimie, mais les différences entre les chimies du carbone et du silicium constituent toujours une question d'actualité, comme d'ailleurs les différences entre les groupes 13 et 14 (principalement entre le bore et le carbone). Les composés de silicium et de germanium ont constitué le sujet d'un article dans Journal of Chemical Education.[28].
Méthodes DFT
Article détaillé : Théorie de la fonctionnelle de la densité.Les méthodes basées sur la théorie de la fonctionnelle de la densité (DFT) sont souvent considérées comme des méthodes ab initio pour la détermination de la structure électronique moléculaire (ou autre), même si les fonctionnelles les plus courantes utilisent des paramètres dérivés de données empiriques, ou de calculs plus complexes. Ceci permet d'affirmer qu'elles peuvent être aussi qualifiée de méthodes semi-empiriques. Il est sans doute plus pertinent de les considérer comme une classe à part. En DFT, l'énergie totale est exprimée en termes dépendant de la densité électronique plutôt qu'en termes de fonctions d'onde. Dans ce type de calculs, il y a un hamiltonien approximé et une expression de la densité électronique totale également approximée. Les méthodes DFT peuvent être extrêmement précises pour un coût de calcul faible. Le défaut majeur est, contrairement aux méthodes ab initio classiques, il n'existe pas de procédé systématique d'amélioration des méthodes par amélioration de la forme de la fonctionnelle.
Méthodes semi-empiriques et empiriques
Article détaillé : Méthodes quantiques semi-empiriques.Les méthodes semi-empiriques de chimie quantique sont basées sur un formalisme Hartree-Fock, mais procèdent à de nombreuses approximations et utilisent des paramètres issus de données empiriques. Elles sont très importantes en chimie numérique pour traiter de grands ensembles moléculaires dans lesquels une méthode Hartree-Fock pure sans approximations est trop coûteuse. L'utilisation de paramètres empiriques peut permettre d'inclure des effets de corrélation dans les méthodes employées.
Les méthodes semi-empiriques succèdent à ce qui est parfois appelé des méthodes empiriques dans lesquelles la partie à deux électrons du hamiltonien n'est pas incluse de manière explicite. Pour les systèmes à électrons π, il s'agit de la méthode de Hückel proposée par Erich Hückel, et pour tous les systèmes d'électrons de valence, la méthode de Hückel étendue proposée par Roald Hoffmann.Mécanique moléculaire
Article détaillé : Mécanique moléculaire.Dans de nombreux cas, les systèmes moléculaires importants peuvent être modélisés sans recourir à des calculs de mécanique quantique intégraux. Les simulations de mécanique moléculaire, par exemple, utilisent une simple expression classique pour l'énergie d'un composé, comme celle de l'oscillateur harmonique. Toutes les constantes nécessaires aux calculs doivent être obtenues à partir de données expérimentales ou de calculs ab initio.
La banque de données sur les composés utilisés pour la paramétrisation - l'ensemble de paramètres résultant étant appelé champ de force - est cruciale pour la fiabilité des calculs en mécanique moléculaire. Un champ de force paramétré pour une classe spécifique de molécules, comme par exemple les protéines, ne sera sans doute pertinent que lorsqu'il sera utilisé pour décrire des molécules de la même classe.Interprétation des fonctions d'ondes moléculaires
Le modèle « Atoms in Molecules » (AIM) de Richard Bader fut développé dans le but de lier de manière effective l'image en mécanique quantique d'une molécule[29] comme fonction d'onde électronique, à des modèles plus anciens mais plus « visibles » chimiquement parlant comme la théorie de la paire de Lewis ou celle de la liaison de valence. Richard Bader a démontré que ces modèles empiriques très utilisés et pratiques sont connectés avec la topologie de la densité de charge quantique. Cette méthode s'améliore sur la base de l'utilisation des charges de Mulliken.
Il existe une autre possibilité de visualisation dans les systèmes chimiques, appelée fonction de localisation électronique, développée par Becke et Edgecombe[30], normalisée (ses valeurs sont comprises en 0 et 1), basée - schématiquement - sur l'expression de la topologie de la densité électronique (et non plus la densité de charge) qui permet de connaître (entre autres) les surfaces d'isodensité et de comparer les « intensités » des liaisons chimiques entre elles.Méthodes de chimie numérique dans les systèmes solides
Les méthodes de chimie numérique peuvent être appliquées à la résolution de problèmes en physique du solide ou assimilés (interfaces, etc.) selon les mêmes approches que pour des systèmes moléculaires, mais avec deux différences remarquables. La première est l'utilisation des différentes symétries spécifiques dans ces ensembles (indiquées par les groupes d'espace) et aussi - et surtout - par un usage intensif des conditions périodiques aux limites. La seconde est la possibilité d'utiliser des fonctions de base complètement délocalisées comme des ondes planes comme alternatives aux fonctions de base centrées sur les atomes des molécules.
La structure électronique d'un cristal est en général décrite par une structure de bande, définissant les énergies des orbitales électroniques pour chaque point de la zone de Brillouin. Les calculs ab initio ou semi-empiriques fournissent les énergies orbitalaires, donc elles peuvent être par conséquent appliquées pour les calculs de structure de bande. On remarquera que s'il est coûteux en temps de calculer l'énergie d'une molécule, il est plus coûteux encore de la calculer pour la liste complète des points de la zone de Brillouin.
Les calculs peuvent utiliser des méthodes Hartree-Fock, post-Hartree-Fock (comme la théorie de la perturbation de Møller-Plesset au second ordre - MP2), et bien sûr la théorie de la fonctionnelle de la densité.Comme pour les molécules, les méthodes employées - générales ou type d'applications au sein d'une application - pour l'étude des systèmes solides dépendent entre autres choses, de la taille des systèmes (influence des lacunes, diffusion, etc.)[31] et des propriétés que l'on cherche à étudier (propriétés mécaniques, comparaisons de stabilité thermodynamique, phonons, conductivité électrique, etc.).
De nombreuses problématiques ont pu émerger de l'utilisation intensive des méthodes de chimie numérique et en particulier des méthodes DFT, ou plus précisément des lacunes constatées dans les approches des sujets et des comparaisons avec les données expérimentales. Ces problématiques touchent à la fois les aspects numériques (progression des algorithmes, parallélisation des codes, etc.), théoriques (introduction et développement de fonctionnelles de la DFT, pseudo-potentiels, etc.) et « expérimentaux » (fiabilité des codes dans leur ensemble, performances machines, etc.). Ainsi, par exemple, une des problématiques majeures de la chimie numérique du solide est le traitement des systèmes comportant une part importante de vide (i.e. cages, interfaces solide-gaz, etc.) par des méthodes d'ondes planes.Exemple : étude de matériaux binaires ou ternaires potentiellement ultra-durs
L'utilisation de calculs intensifs en chimie numérique du solide touche de nombreuses problématiques de cette discipline. On les retrouve dans des domaines aussi variés que la conception de nouveaux matériaux pour batteries, de stockage d'hydrogène, l'étude de nanotubes pour applications de microélectronique ou encore l'étude des comportements de matériaux de type « nucléaires ».
L'une des thématiques abordées par cette recherche ces vingt dernières années porta sur la proposition de nouvelles phases dites ultra-dures afin de pallier l'étroitesse du domaine d'utilisation du diamant dans des procédés de type industriel comme la découpe de matériaux ou autres enrobages de protection (industrie pétrolière). L'un des défauts majeurs montrés par le diamant est sa métastabilité thermodynamique dans les conditions normales de température et pression, sa sensibilité au phénomène d'oxydation et surtout - quand on à faire avec le phénomène d'échauffement par frottement, à sa décomposition au delà de 870 K dans l'air. Bien que connaissant et utilisant le nitrure de bore cubique - moins dur - comme matériau de remplacement dans certains domaines thermodynamiques, l'industrie reste à la recherche de matériaux aux propriétés mécaniques et thermodynamiques pouvant combiner les avantages des deux matériaux.
L'initialisation de cette recherche a réellement eu lieu d'un point de vue théorique dans les années 1980, lorsque les américains A.Y. Liu et M.L. Cohen présentèrent dans un article[32] une équation semi-empirique simple pour formuler de tels matériaux, équation appuyée par des calculs de type ab initio : leur résumé indique « The empirical model indicates that hypothetical covalent solids formed between carbon and nitrogen are good candidates for extreme hardness », ouvrant la voie à la recherche théorique et expérimentale de structures ultra-dures de type carbonitrure (de formule générale CxNy) avec pour prototype C3N4 [33].
Entre cette annonce et l'année 2003 - date de la première publication sur une synthèse de carbonitrure C3N4±δ cristallin tridimensionnel - bien que les équipes d'expérimentateurs n'aient pu que produire quelques phases bidimensionnelles donc impropre à une utilisation comme ultra-durs, les équipes de théoriciens ont proposé un nombre relativement important de structures C3N4 tridimensionnelles susceptibles de remplir les conditions voulues, propositions confortées dans le même temps par des calculs essentiellement de type DFT[34], puis étendu à la recherche d'autres formulations (BC3N3, C11N4, etc.), en interprétant de concert les phénomènes microscopiques en jeu dans la propriété macroscopique de dureté (avec en particulier l'influence de l'existence de doublets électroniques non engagés).Dynamique chimique
Une fois les variables électronique et nucléaire séparées (dans le cas de l'application de l'approximation de Born-Oppenheimer), dans l'approche dépendante du temps, le paquet d'onde correspondant aux degrés de liberté nucléaires est propagé par l'opérateur d'évolution temporelle associé à l'équation de Schrödinger dépendante du temps (pour un hamiltonien du système complet). Dans une approche basée sur la complémentarité de l'énergie, l'équation de Schrödinger dépendante du temps est résolue en utilisant le formalisme de la théorie de la diffusion. Le potentiel représentant les interactions atomiques est donné par les surfaces d'énergies potentielles. En général, ces surfaces sont liées par le biais des termes de couplage vibronique.
Les méthodes les plus courantes pour effectuer la propagation du paquet d'onde associé à la géométrie moléculaire sont :
- la technique de séparation des opérateurs (split operator technique en anglais)
- la méthode multi-configurationnelle dépendante du temps de Hartree (Multi-Configuration Time-Dependent Hartree en anglais, MCTDH)
- la méthode semi-classique
La dynamique moléculaire étudie (moyennant l'utilisation des lois du mouvement de Newton) l'évolution temporelle des systèmes, comprenant les vibrations ou le mouvement brownien, par utilisation d'une description mécanique classique. La combinaison de la dynamique moléculaire avec la théorie de la fonctionnelle de la densité conduit à la méthode de Car et Parrinello.
Liste de codes de chimie numérique
Un certain nombre de codes incluent plusieurs méthodes de chimie quantique, et dans certains cas des méthodes de mécanique moléculaire. Les tableaux suivants - établis en fonction du type de système visé - montrent les capacités des codes les plus communs en indiquant les méthodes utilisées dans une ou plusieurs méthodes.
Codes orientés « systèmes moléculaires »
Code mécanique moléculaire méthodes
semi-empiriquesMéthode
Hartree-FockMéthodes
post-Hartree-FockThéorie de la
fonctionnelle de la densitéACES N N O O N CADPAC N N O O O COLUMBUS N N O O N DALTON N N O O O GAMESS (UK) N O O O O GAMESS (US) Ya O O O O GAUSSIAN O O O O O JAGUAR O N O O O MOLCAS O O O O O MOLPRO N N O O O MPQC N N O O O NWChem O N O O O PLATO O N N N O PQS O O O O O PSI N N O O N Q-Chem O N O O O TURBOMOLE O N O O O a grâce à une interface avec TINKER.
Codes orientés « systèmes cristallins »
Code méthodes
semi-empiriquesMéthode
Hartree-FockMéthodes
post-Hartree-FockThéorie de la
fonctionnelle de la densitéAbinit N N N O ADF N N N O Atomistix Toolkit N N N O CASTEP N N N O CRYSTAL N O N O GAUSSIAN O O O O LMTO N N N O MOPAC O N N N NWChem N O O O PLATO N N N O VASP N N N O WIEN (FPLAPW) N N N O Voir aussi
- Académie internationale de science moléculaire quantique
- Base (chimie quantique)
- Bio-informatique
- Chemo-informatique
- Computational Chemistry List
- Chimie quantique
- Calcul scientifique
- Mécanique statistique
- Physique numérique
- Modélisation moléculaire
- Modélisation moléculaire Monte-Carlo
Notes et références
- ↑ Une simple molécule d'eau (H2O) contient 10 électrons.
- ↑ Et employée dans des simulations de dynamique moléculaire.
- ↑ Un exemple de cette dualité peut être vu dans l'article suivant : E. Betranhandy, S. Matar, « Matériaux ultra-durs : généralités et démarche prospective », dans L'actualité chimique, vol. 294, février 2006, p. 16
- ↑ (en) S.J. Smith, B.T. Sutcliffe, « The development of Computational Chemistry in the United Kingdom », dans Reviews in Computational Chemistry, vol. 70, 1997, p. 271 - 316
- ↑ Henry F. Schaefer III, The electronic structure of atoms and molecules, Addison-Wesley Publishing Co., 1972, 146 p.
- ↑ (en) S.F. Boys, G.B. Cook, C.M. Reeves, I. Shavitt, « Automatic fundamental calculations of molecular structure », dans Nature, no 2, 1956, p. 1207
- ↑ W.G. Richards, T. E. H. Walker et R. K. Hinkley, A bibliography of ab initio molecular wave functions, Clarendon Press
- ↑ (en) H. Preuss, « », dans International Journal of Quantum Chemistry, vol. 2, 1968, p. 651
- ↑ (en) R.J. Buenker, S.D. Peyerimhoff, « », dans Chemical Physics Letters, vol. 3, 1969, p. 37
- ↑ Henry F. III Schaefer, Quantum Chemistry, Clarendon Press
- ↑ A. Streitwieser, J.I. Brauman et C.A. Coulson, Supplementary Tables of Molecular Orbital Calculations, Pergamon Press, 1965
- ↑ John A. Pople, David L. Beveridge, Approximate Molecular Orbital Theory, McGraw Hill, 1970
- ↑ Norman Allinger, « Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms », dans Journal of the American Chemical Society, vol. 99, 1977, p. 8127-8134
- ↑ Computers and Their Role in the Physical Sciences, Routledge 1970
- ↑ Reviews in Computational Chemistry vol 1, preface
- ↑ Une telle recherche est bien sûr dépendante à la fois des coordonnées initiales sur la surface d'énergie potentielle et des algorithmes de recherche de minima.
- ↑ On rappelle que l'équation de Schrödinger n'est analytiquement tractable que pour certains cas minoritaires.
- ↑ C'est-à-dire le mieux qu'il est possible d'obtenir avec une chaîne de caractères de longueur finie sur le calculateur, et dans les approximations mathématiques et physiques effectuées.
- ↑ Mais n'atteignent jamais dans la très grande majorité des cas.
- ↑ Ou, dans d'autres cas, sur les atomes du solide, ou autre.
- ↑ Parfois appelé ansatz.
- ↑ (en) B.T. Colegrove, H.F. Schaefer, « Disilyne (Si2H2) revisited », dans Journal of Physical Chemistry, vol. 94, 1990, p. 5593
- ↑ (en) R.S. Grev, H.F Schaefer, « The remarkable monobridged structure of Si2H2 », dans Journal of Chemical Physics, vol. 97, 1992, p. 7990
- ↑ (en) Zoltán Palágyi, Henry F. Schaefer, Ede Kapuy, « Ge2H2: A Molecule with a low-lying monobridged equilibrium geometry », dans Journal of the American Chemical Society, vol. 115, 1993, p. 6901 - 6903
- ↑ (en) J.C. Stephens, E.E. Bolton, H.F. Schaefer, III, and L. Andrews, « Quantum mechanical frequencies and matrix assignments to Al2H2 », dans Journal of Chemical Physics, vol. 107, 1997, p. 119 - 223
- ↑ (en) Zoltán Palágyi, Henry F. Schaefer III, Ede Kapuy, « Ga2H2: planar dibridged, vinylidene-like, monobridged and trans equilibrium geometries », dans Chemical Physics Letters, vol. 203, 1993, p. 195 - 200
- ↑ Car Al = [Ne] 3s23p1, soit un électron en moins dans les orbitales p par rapport à Si.
- ↑ (en) B.J. DeLeeuw, R.S. Grev, Henry F. Schaefer III, « A comparison and contrast of selected saturated and unsaturated hydrides of group 14 elements », dans Journal of Chemical Education, vol. 69, 1992, p. 441
- ↑ Ou, par extension, d'un autre système chimique comme les solides.
- ↑ (en) A.D. Becke, K.E. Edgecombe, « A simple measure of electron localzation in atomic and molecular-systems », dans Journal of Chemical Physics, vol. 92, 1990, p. 5397-5403
- ↑ Qui peuvent nécessiter une levée de la symétrie cristalline (tridimensionnelle) ou surfacique (bidimensionnelle).
- ↑ (en) A.L. Liu, M.L. Cohen, « Prediction of New Low Compressibility Solids », dans Science, vol. 245, 1989, p. 841
- ↑ Voir sur cette formulation l'article de revue : (en) E. Kroke, M. Schwartz, « Novel group 14 nitrides », dans Coordination Chemistry Reviews, vol. 248, 2004, p. 493
- ↑ Par exemple : (en) D.M. Teter, R.J. Hemley, « Low-Compressibility Carbon Nitrides », dans Science, vol. 271, 1996, p. 53
Autres références non citées
- (en) T. Clark A Handbook of Computational Chemistry, Wiley, New York (1985)
- (en) C. J. Cramer Essentials of Computational Chemistry, John Wiley & Sons (2002)
- (en) R. Dronskowski Computational Chemistry of Solid State Materials, Wiley-VCH (2005)
- (en) F. Jensen Introduction to Computational Chemistry, John Wiley & Sons (1999)
- (en) D. Rogers Computational Chemistry Using the PC, 3rd Edition, John Wiley & Sons (2003)
- (en) A. Szabo, N.S. Ostlund, Modern Quantum Chemistry, McGraw-Hill (1982)
- (en) D. Young Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems, John Wiley & Sons (2001)
- (en) Introduction to Computational Chemistry de David Young
- (en) Cet article est partiellement ou en totalité issu d’une traduction de l’article de Wikipédia en anglais intitulé « Computational chemistry ».
- (en) Cet article est partiellement ou en totalité issu d’une traduction de l’article de Wikipédia en anglais intitulé « Computational chemical methods in solid state physics ».
 Portail de la chimie
Portail de la chimie
Catégorie : Chimie numérique

Wikimedia Foundation. 2010.